Abstract
Methionine is an essential sulfur-containing amino acid that is metabolized mainly in the liver, where it is converted to S-adenosylmethionine (SAM) by methionine adenosyltransferase. Importantly, SAM is a metabolically pleiotropic molecule that participates in three types of biochemical reactions; transmethylation, transsulfuration (which results in the transfer of sulfur from methionine to serine to form cysteine), and amino propylation (to synthesize polyamines). Critical roles of SAM in the liver have been extensively studied using transgenic animals with chronically reduced or increased hepatic SAM levels. Interestingly, both models with abnormal hepatic SAM concentrations develop liver disease suggesting that SAM homeostasis plays a pivotal role in liver disease. The transsulfuration pathway is connected to the production of glutathione (GSH), which has potent antioxidant capacity in the liver. Accumulating data show that GSH depletion renders the liver vulnerable to oxidative stress and prone to progression of liver disease. In this review, we highlight the importance of homeostasis in the metabolism of sulfur-containing amino acids with a particular focus on the transsulfuration pathway which could be a promising therapeutic target in liver injury.
1. INTRODUCTION
Liver disease is considered to be acute or chronic depending on duration of the injury process. Hepatic fibrogenesis is promoted by repeated tissue damage and continues until the damage is repaired. If the injury cannot be repaired during healing, fibrosis will progress to an irreversible stage such as cirrhosis and hepatocellular carcinoma.1) Progression of liver disease is influenced by multiple factors including genetic factors, pathogens, and other environmental influences.2) Accumulating evidence indicates that alterations in the metabolism of sulfur-containing amino acids are important factors contributing to the development of liver disease.
2. METABOLISM OF SULFUR-CONTAINING AMINO ACIDS IN THE LIVER
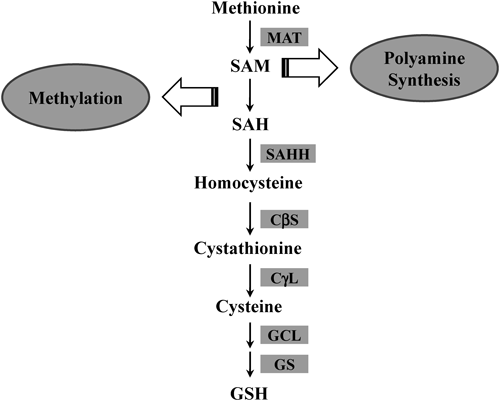
Methionine is an essential sulfur-containing amino acid, and about half of dietary methionine is metabolized in the liver.3) The first step in methionine metabolism is the formation of S-adenosylmethionine (SAM), which is catalyzed by methionine adenosyltransferase (MAT). Mammals have two MAT genes: MAT1A, most highly expressed in normal liver, and ubiquitously expressed MAT2A.4) Most SAM molecules (>85%) generated from methionine are used as methyl donors for transmethylation reactions such as methylation of nucleic acids, phospholipids, histones, biogenic amines, and proteins5) (Fig. 1). In addition to this well-known function, SAM is involved in the synthesis of polyamines, which play important roles in the regulation of transcription, translation, cell proliferation, and apoptosis.6,7) SAM is converted to decarboxylated SAM, which then releases an aminopropyl group used in polyamine synthesis.8) On the other hand, the transsulfuration pathway connects SAM to cysteine biosynthesis and makes SAM an important precursor of glutathione (GSH); the transsulfuration pathway is especially active in the liver (Fig. 2). S-Adenosylhomocysteine (SAH), the byproduct of SAM in transmethylation reactions, is hydrolyzed into homocysteine and adenosine by S-adenosylhomocysteine hydrolase (SAHH). Homocysteine is the substrate for two competitive reactions, remethylation and transsulfuration, in the metabolism of sulfur-containing amino acids in the liver.9) Remethylation of homocysteine is mediated by methionine synthase (MS) or betain-homocysteine methyltransferase (BHMT), and is followed by the generation of methionine. The transsulfuration pathway metabolizes homocysteine into cysteine via cystathionine β-synthase (CβS) and cystathionine γ-lyase (CγL). Subsequently, cysteine is metabolized into taurine, inorganic sulfate, or GSH in the liver.10) In this article, we review the importance of the transsulfuration pathway with a particular focus on the role of SAM and GSH in liver injury and regeneration.
3. TRANSSULFURATION IN LIVER INJURY
Both acute and chronic liver injury are closely related to the loss of homeostasis in the hepatic metabolism of sulfur-containing amino acids, which is caused by the deregulation of metabolic enzymes. In agreement with this, deletion of MAT1A, or BHMT, or CβS in mice caused fatty liver disease; in particular, MAT1A-deficient mice spontaneously developed hepatocellular carcinoma.
3.1. Alteration of SAM Levels in Liver InjuryVarious pathological conditions that promote oxidative and/or nitrosative stress, such as alcohol consumption, viral hepatitis, septic shock, and toxin exposure, may inactivate MAT1A.11) Notably, patients with hepatic cirrhosis have been shown to have diminished SAM biosynthesis because of reduced expression and activity of MAT1A.12) Considering that SAM is a biochemical intermediate in GSH synthesis, the reduced SAM level will further down-regulate the hepatocellular defense against oxidative stress and worsen liver injury.
The effects of chronic SAM depletion have been extensively studied in MAT1A and BHMT knockout mice. MAT1A-deficient mice showed increased susceptibility to steatosis and oxidative liver injury, as well as spontaneous steatohepatitis and hepatocellular carcinoma.13) In a BHMT knockout mouse, chronically reduced SAM levels were accompanied by severe hyperhomocysteinemia.14) This mouse showed reduced methylation potential and phosphatidylcholine level, decreased very low density lipoprotein (VLDL) secretion, and increased hepatic triglyceride accumulation. Finally, similarly to a MAT1A knockout mouse, this mouse developed hepatocellular carcinoma. The effects of chronically elevated SAM levels have been studied in mice lacking glycine N-methyltransferase (GNMT).15) GNMT is one of the methyltransferases that normally maintain the SAM-to-SAH ratio by using excess SAM to methylate glycine to sarcosine. Interestingly, these mice also developed liver steatosis, fibrosis, and hepatocellular carcinoma. Collectively, these findings in MAT1A, BHMT, and GNMT knockout mice emphasize that maintaining the homeostasis of hepatic SAM levels is critical for the prevention of liver injury.
3.2. Changes in GSH in Liver InjuryGSH is the most abundant non-protein thiol composed of three amino acids, glutamine, cysteine, and glycine. The cysteine residue with its thiol group is a potent reductant and provides defense against oxidative stress.16) GSH is also a key molecule in redox signaling and detoxification of xenobiotics, and regulates cell survival, immune function, and fibrogenesis.17) There is increasing evidence that dysregulation of GSH synthesis contributes to the pathogenesis of many liver diseases including non-alcoholic fatty liver disease, alcoholic liver disease, and drug-induced liver injury.18)
GSH synthesis is mediated by glutamate cysteine ligase (GCL), which is composed of a catalytic (GCLC) and a modifier (GCLM) subunit, and GSH synthase (GS), which catalyze consecutive reactions. The major factors affecting GSH synthesis are the availability of the substrate, cysteine, and the activity of GCL, which is the rate-limiting enzyme.16) The key transcription factors that regulate the expression of genes encoding GCLC, GCLM, and GS include NF-E2-related factor 2 (Nrf2), activating protein-1 (AP-1), and nuclear factor kappa B (NF-κB).19,20) Beyond decreased metabolic substrates of GSH synthesis via down regulation of SAM level, various factors can modulate GSH biosynthesis and expenditure followed by a reduction in antioxidant capacity in liver injury.
Endotoxemia, which is frequently observed in cirrhotic patients, correlates with the degree of liver failure and worsens liver diseases such as alcohol-induced liver injury and non-alcoholic steatohepatitis.21,22) Lipopolysaccharide (LPS) challenge in mice lowered the hepatic GSH level and induced liver injury. However, exogenous GSH or betaine, which increased the SAM level, suppressed the LPS-induced systemic inflammatory response and liver injury.23,24) A recent study has shown that the LPS-induced reduction in GSH coincides with a marked reduction in the expression of GSH synthetic enzymes, GCL and GS, at both the mRNA and protein levels.25)
Alcoholic liver injury is a major cause of chronic liver disease. In particular, alcohol-induced oxidative stress appears to play a central role in pathways involved in initiation and progression of alcoholic liver disease.26) Considering the potent antioxidant capacity of GSH, many investigators have focused on the regulation of GSH synthesis and suggested that alterations in hepatic transsulfuration are important for the development of alcoholic liver injury.27) Patients with alcoholic liver disease have low hepatic and plasma GSH levels due to multiple factors that impair cysteine availability, such as oxidative stress, nutritional deficiency, and abnormalities in the transsulfuration pathway.28) In addition, patients hospitalized for alcoholic hepatitis showed a 50% reduction in GCLC and GS mRNA levels as compared with normal liver controls.28)
Hepatic fibrosis, which occurs in most types of chronic liver disease, represents excessive accumulation of extracellular matrix including collagen. trans-Differentiation of quiescent hepatic stellate cells (HSCs) to myofibroblasts is mediated by inflammatory cytokines and reactive metabolites released from damaged hepatocytes and activated Kupffer cells, followed by synthesis of pro-fibrogenic factors.29) Recently, it has been suggested that a reduced GSH level promotes activation of HSCs, leading to advanced liver injury. Indeed, a lower hepatic GSH level greatly potentiated bile duct ligation-induced fibrosis.30) On the other hand, preventing a decrease in hepatic GSH ameliorated hepatic fibrosis.31) Epigallocatechin gallate and curcumin showed an anti-fibrotic effect in HSCs by maintaining GSH levels.32,33)
4. IMPORTANCE OF THE TRANSSULFURATION PATHWAY IN LIVER REGENERATION
The liver is a special organ capable of regeneration following physical or chemical tissue injury. Injured tissues or organs are able to repair, but only the liver possesses the capability of compensatory growth to replenish lost functional volume.34) Experimentally, partial hepatectomy is a standard model to study the mechanisms of recovery from liver injury. It is suggested that liver regeneration is accompanied by a profound change in the metabolites of the transsulfuration pathway in the liver.35) However, the physiological role of this pathway in liver regeneration is still unclear. To clarify the importance of the transsulfuration pathway during liver regeneration, a number of studies have used chemical inhibitors or knockout mice together with the partial hepatectomy model.
In the early phase of liver regeneration, an increase in the GSH level is a common phenomenon and it has been suggested that GSH may be essential for liver regeneration.36) However, studies that used two compounds, buthionine-sulfoximine (BSO) and phorone (PHO), to deplete GSH showed different results depending on the mechanism of action of these compounds.37) BSO induces the depletion of both SAM and GSH via inhibiting their synthetic enzymes, MAT and GCL, respectively. In regenerating rat livers, administration of BSO was shown to reduce liver regeneration as evidenced by a decrease in DNA synthesis; this effect of BSO was prevented by SAM administration, but was rather exacerbated by GSH treatment.38) On the other hand, PHO challenge, which depletes GSH by conjugation with GSH and increases SAM synthesis via induction of MAT activity, enhanced regeneration, suggesting that the functions of SAM and GSH in liver regeneration could be different.37)
Although chemical inhibitors of a metabolic pathway can provide useful mechanistic information, transient manipulation and potential off-target effects limit the use of inhibitors for uncovering the roles of individual metabolites. Therefore, some investigators used the knockout approach to overcome these concerns. Mice lacking MAT1A have chronic hepatic SAM deficiency and impaired liver regeneration after partial hepatectomy.39) Interestingly, MAT1A knockout hepatocytes had higher baseline of DNA synthesis than that of wild-type hepatocytes but failed to respond to the mitogenic effect of hepatocyte growth factor, suggesting that chronic depletion of hepatic SAM results in a loss of responsiveness to mitogenic signals followed by abnormal regeneration. On the other hand, the regenerating livers of Gclm−/− mice, which have consistent depletion of GSH, showed an overall delay in cell cycle progression with slower DNA synthesis, mitosis, and expression of cell cycle proteins, accompanied by a delay in the expression of downstream targets of NF-κB.40) This study suggested that GSH plays a significant role in hepatic NF-κB activation, which is necessary for normal liver regeneration.
5. CONCLUDING REMARKS
Metabolism of sulfur-containing amino acid is involved in the formation of various metabolites that play important physiological roles. Impairment of this metabolism is strongly associated with chronic liver disease and also causes the failure of liver regeneration after tissue damage. Conversely, modification of abnormality in hepatic metabolism of sulfur-containing amino acid has been effective against liver injury. Finally, it is suggested that approaches to maintaining homeostasis in hepatic metabolism of sulfur-containing amino acid could be a promising target in the therapy of liver disease.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2014R1A1A1005435) and Pusan National University Research Grant 2013.
Conflict of Interest
The author declares no conflict of interest.
REFERENCES
- 1) Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc., 55, 434–438 (1980).
- 2) Bataller R, North KE, Brenner DA. Genetic polymorphisms and the progression of liver fibrosis: a critical appraisal. Hepatology, 37, 493–503 (2003).
- 3) Finkelstein JD. Methionine metabolism in mammals. J. Nutr. Biochem., 1, 228–237 (1990).
- 4) Lu SC, Mato JM. S-Adenosylmethionine in liver health, injury, and cancer. Physiol. Rev., 92, 1515–1542 (2012).
- 5) Lieber CS, Packer L. S-Adenosylmethionine: molecular, biological, and clinical aspects—an introduction. Am. J. Clin. Nutr., 76, 1148S–1150S (2002).
- 6) Perez-Leal O, Merali S. Regulation of polyamine metabolism by translational control. Amino Acids, 42, 611–617 (2012).
- 7) Jänne J, Alhonen L, Leinonen P. Polyamines: from molecular biology to clinical applications. Ann. Med., 23, 241–259 (1991).
- 8) Lu SC. S-Adenosylmethionine. Int. J. Biochem. Cell Biol., 32, 391–395 (2000).
- 9) Stead LM, Brosnan ME, Brosnan JT. Characterization of homocysteine metabolism in the rat liver. Biochem. J., 350, 685–692 (2000).
- 10) Kim SK, Kim YC. Effects of betaine supplementation on hepatic metabolism of sulfur-containing amino acids in mice. J. Hepatol., 42, 907–913 (2005).
- 11) Mato JM, Corrales FJ, Lu SC, Avila MA. S-Adenosylmethionine: a control switch that regulates liver function. FASEB J., 16, 15–26 (2002).
- 12) Duce AM, Ortiz P, Cabrero C, Mato JM. S-Adenosyl-L-methionine synthetase and phospholipid methyltransferase are inhibited in human cirrhosis. Hepatology, 8, 65–68 (1988).
- 13) Martínez-Chantar ML, Corrales FJ, Martínez-Cruz LA, García-Trevijano ER, Huang ZZ, Chen L, Kanel G, Avila MA, Mato JM, Lu SC. Spontaneous oxidative stress and liver tumors in mice lacking methionine adenosyltransferase 1A. FASEB J., 16, 1292–1294 (2002).
- 14) Teng YW, Mehedint MG, Garrow TA, Zeisel SH. Deletion of betaine-homocysteine S-methyltransferase in mice perturbs choline and 1-carbon metabolism, resulting in fatty liver and hepatocellular carcinomas. J. Biol. Chem., 286, 36258–36267 (2011).
- 15) Martínez-Chantar ML, Vázquez-Chantada M, Ariz U, Martínez N, Varela M, Luka Z, Capdevila A, Rodríguez J, Aransay AM, Matthiesen R, Yang H, Calvisi DF, Esteller M, Fraga M, Lu SC, Wagner C, Mato JM. Loss of the glycine N-methyltransferase gene leads to steatosis and hepatocellular carcinoma in mice. Hepatology, 47, 1191–1199 (2008).
- 16) Meister A, Anderson ME. Glutathione. Annu. Rev. Biochem., 52, 711–760 (1983).
- 17) Ookhtens M, Kaplowitz N. Role of the liver in interorgan homeostasis of glutathione and cyst(e)ine. Semin. Liver Dis., 18, 313–329 (1998).
- 18) Chen Y, Dong H, Thompson DC, Shertzer HG, Nebert DW, Vasiliou V. Glutathione defense mechanism in liver injury: insights from animal models. Food Chem. Toxicol., 60, 38–44 (2013).
- 19) Wild AC, Moinova HR, Mulcahy RT. Regulation of gamma-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J. Biol. Chem., 274, 33627–33636 (1999).
- 20) Yang H, Magilnick N, Ou X, Lu SC. Tumour necrosis factor alpha induces co-ordinated activation of rat GSH synthetic enzymes via nuclear factor kappaB and activator protein-1. Biochem. J., 391, 399–408 (2005).
- 21) Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am. J. Physiol. Gastrointest. Liver Physiol., 283, G256–G265 (2002).
- 22) Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc. Natl. Acad. Sci. U.S.A., 94, 2557–2562 (1997).
- 23) Kim SK, Kim YC. Attenuation of bacterial lipopolysaccharide-induced hepatotoxicity by betaine or taurine in rats. Food Chem. Toxicol., 40, 545–549 (2002).
- 24) Sun S, Zhang H, Xue B, Wu Y, Wang J, Yin Z, Luo L. Protective effect of glutathione against lipopolysaccharide-induced inflammation and mortality in rats. Inflamm. Res., 55, 504–510 (2006).
- 25) Tomasi ML, Ryoo M, Yang H, Iglesias Ara A, Ko KS, Lu SC. Molecular mechanisms of lipopolysaccharide-mediated inhibition of glutathione synthesis in mice. Free Radic. Biol. Med., 68, 148–158 (2014).
- 26) Kim SJ, Lee JW, Jung YS, Kwon do Y, Park HK, Ryu CS, Kim SK, Oh GT, Kim YC. Ethanol-induced liver injury and changes in sulfur amino acid metabolomics in glutathione peroxidase and catalase double knockout mice. J. Hepatol., 50, 1184–1191 (2009).
- 27) Kim SK, Seo JM, Jung YS, Kwak HE, Kim YC. Alterations in hepatic metabolism of sulfur-containing amino acids induced by ethanol in rats. Amino Acids, 24, 103–110 (2003).
- 28) Lee TD, Sadda MR, Mendler MH, Bottiglieri T, Kanel G, Mato JM, Lu SC. Abnormal hepatic methionine and glutathione metabolism in patients with alcoholic hepatitis. Alcohol. Clin. Exp. Res., 28, 173–181 (2004).
- 29) Bataller R, Brenner DA. Liver fibrosis. J. Clin. Invest., 115, 209–218 (2005).
- 30) Ramani K, Tomasi ML, Yang H, Ko K, Lu SC. Mechanism and significance of changes in glutamate-cysteine ligase expression during hepatic fibrogenesis. J. Biol. Chem., 287, 36341–36355 (2012).
- 31) Yang H, Ko K, Xia M, Li TW, Oh P, Li J, Lu SC. Induction of avian musculoaponeurotic fibrosarcoma proteins by toxic bile acid inhibits expression of glutathione synthetic enzymes and contributes to cholestatic liver injury in mice. Hepatology, 51, 1291–1301 (2010).
- 32) Fu Y, Zheng S, Lu SC, Chen A. Epigallocatechin-3-gallate inhibits growth of activated hepatic stellate cells by enhancing the capacity of glutathione synthesis. Mol. Pharmacol., 73, 1465–1473 (2008).
- 33) Zheng S, Yumei F, Chen A. De novo synthesis of glutathione is a prerequisite for curcumin to inhibit hepatic stellate cell (HSC) activation. Free Radic. Biol. Med., 43, 444–453 (2007).
- 34) Michalopoulos GK, DeFrances MC. Liver regeneration. Science, 276, 60–66 (1997).
- 35) Jung YS, Kim SJ, Kwon Y, Kim YC. Kwon do Y, Kim YC. Metabolomic analysis of sulfur-containing substances and polyamines in regenerating rat liver. Amino Acids, 42, 2095–2102 (2012).
- 36) Huang ZZ, Li H, Cai J, Kuhlenkamp J, Kaplowitz N, Lu SC. Changes in glutathione homeostasis during liver regeneration in the rat. Hepatology, 27, 147–153 (1998).
- 37) Jung YS, Kim SJ, Kwon DY, Jun DS, Kim YC. Significance of alterations in the metabolomics of sulfur-containing amino acids during liver regeneration. Biochimie, 95, 1605–1610 (2013).
- 38) Holecek M, Skopec F, Sprongl L. Influence of buthionine sulfoximine, S-adenosylmethionine and glutathione on liver regeneration following partial hepatectomy. Arzneimittelforschung, 50, 1093–1098 (2000).
- 39) Chen L, Zeng Y, Yang H, Lee TD, French SW, Corrales FJ, Garcia-Trevijano ER, Avila MA, Mato JM, Lu SC. Impaired liver regeneration in mice lacking methionine adenosyltransferase 1A. FASEB J., 18, 914–916 (2004).
- 40) Riehle KJ, Haque J, McMahan RS, Kavanagh TJ, Fausto N, Campbell JS. Sustained glutathione deficiency interferes with the liver response to TNF-alpha and liver regeneration after partial hepatectomy in mice. J. Liver Disease Transplant., 1 (2013).