Current Topics: Cell Death in Neuromuscular Diseases
Choose Delicately and Reuse Adequately: The Newly Revealed Process of Autophagy
2015 Volume 38 Issue 8 Pages 1098-1103
Details
2015 Volume 38 Issue 8 Pages 1098-1103
Autophagy is a degradation system for intracellular components. One of the roles of autophagy is the prompt removal of damaged organelles. Another unique role is to supply resources that maintain metabolism in response to the cellular nutritional state. Precise management of all the components in the autophagic system is essential for cellular health. Especially important are the selectivity of target cargos for autophagy, and the coordination of autophagy with the lysosomal catabolic process. This review outlines our current understanding of autophagy and discusses potential therapeutic perspectives. Emphasis will be given to lysosomal function as a central controller of metabolism, and to selective autophagy as a key mechanism for the efficient removal of dysfunctional organelles.
Autophagy is a degradation process that digests intracellular components via lysosomes. The autophagy–lysosome system is responsible for eliminating obsolete long-lived proteins and organelles which are damaged, dysfunctional or aged. Since lysosomes break down cellular wastes into the resources of metabolism, such as amino acids and free fatty acids for recycling, autophagy consequently plays a role in maintaining cellular metabolism. Since autophagy also functions in the control of the quality of cellular components, it is extremely important in maintaining cellular homeostasis. Although autophagy is vital for cell survival, under certain circumstances it is also implicated in cell death.1,2) Thus, autophagy appears to be a double-edged sword that could be either protective or detrimental. Numerous studies have suggested autophagy as a potential therapeutic target.3–5) More than thirty autophagy-related (Atg) proteins have been identified in the last decade. Autophagy is highly regulated at multiple steps. It is necessary to understand exactly how each step along the autophagy–lysosome pathway is regulated before any therapeutic strategies can be designed to modulate autophagic activity for promoting either cell survival or cell death under different pathological conditions. This review will outline the autophagy pathway focusing on recent findings regarding the mechanisms of selective autophagy, as well as the role of lysosomes in the overall control of autophagy.
Autophagy is a lysosomal degradation system by which cells can eliminate and recycle surplus proteins and damaged organelles.6,7) Currently, four types of autophagy have been reported in mammals, including macroautophagy, alternative autophagy, microautophagy and chaperone-mediated autophagy (CMA). These are identified by differences in their observational characteristics and/or required factors.
2.1. MacroautophagyMacroautophagy is the most extensively studied and best characterized process among all types of autophagy. In this process, double-membrane vesicles named “autophagosomes” sequestrate cytoplasmic constituents and then fuse with lysosomes, where the lysosomal acidic hydrolases then break down the constituents into amino acids, free fatty acids and nucleic acids. The formation of autophagosomes is separated into three steps: phagophore formation, elongation and sequestration. A number of factors play roles in each step of this autophagic process, including autophagosome formation, fusion with the lysosome and degradation within the lysosome. Phagophore formation is the initial step. This membrane isolation process occurs at the phagophore assembly site (PAS) and is regulated by the activity of class III phosphatidylinositol 3-kinase (PI3K). PI3K exists in a complex that also includes Beclin1 (Atg6), Vps15 and Vps34. Beclin1 activity is regulated by various binding proteins. Ambra1, UV radiation resistance associated gene protein (UVRAG), and bif-1 induce autophagosome formation when they bind to Beclin1. Early endosomal guanosine 5′-triphosphatase (GTPase) Rab5 binds to Vps34 and induces autophagy.8,9) In contrast, anti-apoptotic proteins Bcl-2 and Bcl-XL bind to and sequester Beclin1 from the complex, thereby inhibiting autophagy.10,11) Beclin1 is a point of crosstalk between autophagy and apoptosis. In response to certain apoptotic stimuli, Beclin1 is cleaved by Caspase-3, and the carboxyl-terminal fragment localizes on the mitochondria to promote apoptosis.11–13)
ULK1 (Atg1) complex is also a well-characterized regulator in this initiation step. ULK1, ATG13 and FIP200 are the main components of this complex. This complex is described as a mediator of nutrient signaling that regulates the rate of autophagy. The protein kinase ULK1 has multiple functions in autophagy regulation. ULK1 phosphorylates FIP200 to induce autophagosome formation. ULK1 can also phosphorylate Beclin1 at Ser 14 to activate the PI3K complex.14) The activity of ULK1 is regulated by phosphorylation, but different sites of phosphorylation have different consequences. AMP activated protein kinase (AMPK) is a key energy sensor responding to low ATP levels. AMPK promotes autophagy through the phosphorylation of ULK1 at Ser 317 and Ser 777. Conversely, a mammalian target of rapamycin (mTOR), a central cell-growth regulator that integrates growth factor and nutrient signals, inhibits autophagy by phosphorylating ULK1 at Ser 757, thereby disrupting the interaction between ULK1 and AMPK.15) Thus, ULK1 phosphorylation is responsible for maintaining energy and nutrient homeostasis by matching the level of autophagy and the levels of intracellular ATP, amino acids and carbohydrates.16) One study has shown that baicalein leads to human cancer cell death by inducing autophagy via AMPK/ULK1 signaling activation.17) On the other hand, reactive oxygen species (ROS) inhibit autophagy by downregulating the transcription of ULK1 via p70S6K/p53 signaling in selenite-treated NB4 cells.18) These results suggest that physiological or pathological consequences of autophagy depend on the context, such as environments, organs, and stimuli.
Following the autophagosome membrane assembly, membranes continue to elongate and to sequester intracellular components. Two ubiquitin-like conjugation systems, Atg8/MAP-LC3/GABARAP/GATE-16 and Atg5/Atg12/Atg7, play important roles in the elongation process. LC3 is cleaved by Atg4 to expose a carboxyl terminal glycine (LC3-I). Through activation by the E1-like enzyme Atg7 followed by covalent attachment to a phosphatidylethanolamine molecule by an E2-like enzyme Atg3, LC3-I is converted to LC3-II, which is the form to be recruited into the phagophore membrane upon the induction of autophagy. A covalently conjugated Atg5–Atg12–Atg16 complex is required for the lipidation and recruitment of LC3-II in the expanding phagophore. Another autophagy protein, Atg9 also plays a key role in this process. Atg9 is a transmembrane protein prominently located at the trans-Golgi network (TGN) under nutrient-rich conditions, and it can travel among multiple peripheral sites including the PAS. With this feature, Atg9 has been considered part of the system essential for phagophore expansion. Indeed, Atg9-deficient cells cannot complete autophagosome formation responding to starvation.19,20) The activation of the AMPK-ULK1 signaling pathway promotes the localization of Atg9 to autophagosomes,21) suggesting that Atg9 is a nutrient-sensitive regulator in the autophagy process. The transport of Atg9 between the trans-Golgi membrane and phagophore is also regulated by multiple Atg proteins. Efficient anterograde delivery involves Atg9 transport factors, such as Atg23 and Atg27, whereas retrograde movement away from the PAS requires the Atg1 kinase complex, Atg18 and Atg2.22)
The maturation of autophagosomes or endosomes is an essential process for completing the fusion of the autophagosome with the lysosome. The membrane trafficking small Ras-like GTPase Rab proteins are in charge of the vesicle trafficking process. These proteins regulate not only early or late endosome formation and vesicle fusion, but also autophagosome formation and fusion with the lysosome. Indeed, multiple Rab proteins are involved in various stages of autophagy. Rab1, Rab5, Rab7, Rab9A, Rab11, Rab23, Rab32, and Rab33B all participate in autophagosome formation. Rab7, Rab8B, and Rab24 have key roles in the maturation of autophagosomes,23) which then fuse with lysosomes to form autolysosomes. Recently, it has been reported that a soluble NSF attachment protein receptor (SNARE) complex consisting of syntaxin 17 (Stx17), soluble NSF attachment protein (SNAP)-29 and vesicle-associated membrane protein 8 (VAMP8) plays a role in autophagosome maturation, thereby promoting autophagosome–lysosome fusion.24) Since mTOR complex activity is dependent on specific GTPases, and the Rab proteins mentioned above are GTPases, the manipulation of G-protein could be a potent target for autophagy regulation. Activating G-protein Signaling 3 (AGS3) protein is highly expressed in the brain and contributes to cardiac and metabolic functions.25) Interestingly, both the overexpression and down-regulation of AGS3 induces autophagy,26) confirming the necessity of fine-tuning in autophagy when developing therapeutic strategies.
2.2. Alternative AutophagyIn the above-described canonical autophagy, the formation of autophagosome requires LC3, ATG5 and ATG7. However, Nishida et al. showed that there exists a non-canonical pathway that leads to the formation of autophagosomes without requiring LC3, ATG5 or ATG7.27) This pathway relies on Rab9 and is still Beclin1 dependent. Furthermore, it has been reported that there is another Beclin1-independent, but ATG7-dependent pathway.28) In addition, another peptidase-resistant peptide degradation pathway has been reported to depend on Beclin1 but not ATG5/ATG7. Therefore, currently, there are at least four distinguishable systems that can lead to autophagosome formation.28) Past research has focused on the physiological and pathological roles of canonical macroautophagy in health and disease. Future studies should also elucidate the functional roles of these newly identified non-canonical autophagic pathways.
2.3. Microautophagy and Chaperone-Mediated Autophagy (CMA)The uniqueness of microautophagy and CMA is that these processes do not require autophagosome formation for the delivery of targeted cargos to lysosomes. However, the molecular mechanisms that mediate microautophagy and CMA are not fully understood.
While macroautophagy is generally considered a non-selective proteolytic process, microautophagy is mediated by direct lysosomal engulfment of the cytoplasmic cargo, and could be non-selective or selective for targeting organelles to the lysosome.29,30) However, CMA is believed to be a selective mechanism. With cooperation of the cytosolic chaperone Hsc70, other co-chaperones and the lysosomal receptor LAMP2A, proteins are delivered directly to the lysosomal lumen and degraded for intracellular quality control. The substrate requires a KFERQ peptide motif to be recognized by Hsc70. Although the effect of CMA on those proteins is limited, it is suggested that the impairment of CMA is tightly linked to the pathogenesis of neurodegenerative diseases and cancer,31) indicating that CMA is an indispensable pathway in maintaining health.
Lysosomes are in charge of the final stage of autophagy. The organelle has an acidic lumen containing over 60 different hydrolases for digesting cellular components. Recent studies suggest that the lysosome is not just a garbage disposal and recycling station; it is also a traffic control center of metabolism under stress, and is involved in repairing injured plasma membranes.32) The physiological roles of the lysosome have been defined from studies of various diseases in which the lysosome is dysfunctional. Lysosome Storage Disorders (LSD) are a group of about 50 diseases characterized by an accumulation of incomplete byproducts in waste processing,33) resulting in the formation of a large vacuole. Single or multiple lysosomal gene mutations can cause LSD, suggesting that the lysosome consists of few redundant proteins, and its normal function relies on delicate regulation in the loading and processing of materials.
It has also been known that lysosomal membrane permeabilization (LMP) leads to the leakage of lysosomal enzymes into the cytosol.34,35) LMP is a potentially lethal event. This event occurs prior to, or as a signature of, lysosomal dysfunction in response to excessive stress. Once lysosomal proteases such as Cathepsin B/D/L (CTSB/D/L) are leaked into the cytosolic or extracellular compartment, they may lead to cell death by the digestion of vital proteins and the activation of caspases.34) It has been reported that there are ROS-dependent and -independent LMPs, suggesting that multiple signaling pathways converge on this event to transduce the signal.34)
The molecular mechanisms mediating lysosomal signaling have been gradually unveiled. Lysosomal nutrient sensor (LYNUS) machinery consists of the mTORC1 complex, Ragulator, and lysosomal V-ATPase. This transduces the signal of lysosomal nutrient levels via transcription factor EB (TFEB) phosphorylation.32,36) When TFEB is phosphorylated, it is dissociated from the LYNUS complex and translocates into the nucleus to activate the transcription for both Atg genes and lysosomal genes, resulting in the activation of autophagy and lysosomal biogenesis. Through this system, the lysosome regulates growth signaling pathways and autophagy with respect to the nutrient status. Thus, the lysosome is not only the waste disposal site, processing a large amount of cellular components, but also plays an important role as a control center of cellular metabolism. Thus, manipulating lysosomal activity and function may be an effective therapeutic strategy for maintaining cellular homeostasis under certain conditions.36,37)
Autophagy has primarily been described as a non-selective dynamic process to eliminate bulky garbage or to reuse metabolic resources under nutrient stressed conditions. However, recent studies demonstrate that intracellular organelles are selectively removed by autophagy. These processes are collectively termed organellophagy.38) Selective autophagy has been described for mitochondria (mitophagy),39) peroxisomes (pexophagy),40) lysosomes (lysophagy),41) aggresomes (aggrephagy),7,42) liposomes (lipophagy),43) the nucleus (nucleophagy),44,45) ribosomes (ribophagy),46) endoplasmic reticulum (ERphagy),47,48) and even DNA (DNautophagy)49) and RNA (RNautophagy).50) Selective autophagy allows the cell to efficiently control the quality and quantity of organelles without causing excessive removal. In addition, it can selectively provide required metabolic resources to meet demands, such as promoting protein de novo synthesis (ribophagy), enhancing beta-oxidation (lipophagy), and enhancing the turnover of nucleic acids (DN-/RN-autophagy). There are two molecular mechanisms underlying target recognition. In the receptor-mediated process, Atg8 family-interacting motif (AIM)-containing receptors are placed on the surface of the organelle, and interact directly or indirectly with the adaptor proteins Atg8 (LC3, GABARAP, and GATE-16) so that the autophagosomes can recognize their cargos for sequestration. The other is a ubiquitin-mediated process. Ubiquitin E3 ligases are involved in this process. Under stress or signaling activation, the peripheral and/or membrane-anchored proteins on the surface of cargos are tagged with polyubiquitin.51,52) Cytosolic adaptors, such as p62 and NBR1, recognize these ubiquitin chains. These adaptors contain an LC3 interaction domain which can recruit autophagosomes to the target site. Both processes are regulated by multiple protein kinases. Recent studies have explored the regulators of organellophagy, especially mitophagy. Mitochondria are the platform of ATP synthesis. Because of the mitochondria’s role as the energy plant, it is easily exposed to and damaged by oxidative stress. Damaged or otherwise dysfunctional mitochondria are eliminated through the autophagy–lysosome pathway. This process is referred to as mitophagy. When mitophagy is induced, injured mitochondria are tagged with ubiquitin by an E3 ubiquitin ligase such as Parkin, Smurf1, or March5. The tagged mitochondria are recruited by p62 or HDAC6 and then sent to the autophagosome. Mitochondria also have ubiquitin independent LC3 receptor proteins such as BNIP3, NIX and FUNDC1.53,54)
Non-selective macroautophagy and selective organellophagy can be regulated independently. This allows cells to preserve or remove organelles depending on the demands, regardless of the level of overall autophagic flux. Indeed, pexophagy is induced upon a shift from starvation to nutrient-rich media,55,56) while the shift itself can inhibit macroautophagy. Also, macroautophagy is inhibited in the type 1 diabetic mouse heart, which is protective. However, this reduced autophagy was associated with partially restored mitophagy and the increased expression and mitochondrial localization of Rab9, an essential regulator of alternative autophagy.57) Thus, even if general macroautophagy is reduced in a diseased state, other more selective autophagy may remain normal or even enhanced. Moreover, selective autophagy may use different mechanisms to target an organelle for degradation. For example, it has been reported that there are at least three types of mitophagy.58) Type 1 mitophagy is induced by nutrient deprivation in which Beclin1 activation leads to the formation of a cup-shaped phagophore that sequesters a mitochondrion in coordination with mitochondrial fission. Type 2 mitophagy is triggered by mitochondrial damage in which LC3-incorporated membrane vesicles attach to the mitochondrion and coalesce to form mitophagosome in a Beclin1-independent manner. The type 3 mitophagy process is like microautophagy in which mitochondria become internalized into multivesicular bodies in a Pink1/Parkin-dependent fashion, and subsequently fuse with lysosomes.
As described above, autophagy is precisely controlled by the orchestrated function of many different proteins in response to stimuli received at multiple points throughout the autophagy–lysosome pathway (Fig. 1). Starvation is the most extensively studied autophagy inducer. A recent study has revealed that starvation promotes the fusion of autophagosomes and lysosomes by the downregulation of O-linked N-acetylglucosamine modification of SNAP-29.59) Thus, cargo segregation by the autophagosome and its catabolism by the lysosome seem well orchestrated under a poor nutrient environment. In contrast, the promotion of cytoprotective autophagy for the removal of dysfunctional organelles can be easily unleashed under pathological conditions. Additionally, the basal demands for autophagy in cellular quality control are different among organs or cell types. Thus, manipulating autophagy can be either protective or detrimental depending upon the cell types and/or stress conditions. To harness autophagy for therapy, the cargo selection mechanisms are potential targets for manipulation. Since most types of organellophagy seem to share a ubiquitylation mechanism, it is important to identify the organelle specific ubiquitin E3 ligases involved in each pathological condition. Recent studies suggest that multiple metabolic indices can be checkpoints of cell death.60) In view of maintaining metabolic homeostasis, it is also important to manage the capacity of lysosomal degradation. TFEB is a potential candidate to be targeted, since it governs the signal transduction converging on lysosomes, wherein lysosomal turnover and autophagy are regulated. Prior to developing an intervention for diseases in which the autophagy–lysosome pathway is involved, one should first identify the mechanisms underpinning the disease process.
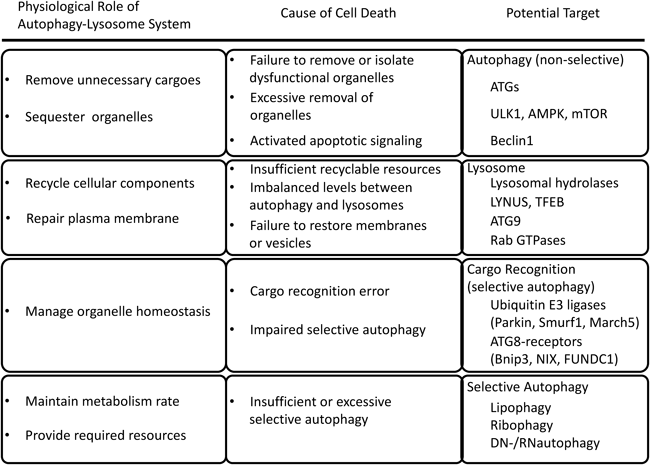
Although the central roles and key features of autophagy have been well characterized, new roles and mechanisms of this degradation and recycling system continue to emerge. Comprehensive approaches are particularly important for fully understanding the precise mechanisms of regulation in the context of individual stresses, and under specific physiological or pathological conditions. A number of intriguing questions remain to be addressed. Are there any other types of autophagy or new players? Does organellophagy involve multiple types of autophagy? If so, how are these types selected under certain conditions? What kinds of stress can trigger lysosomal membrane permeabilization? What are the physiological or pathological roles of lysosomal signaling, and what is its involvement in human disease?
Further, the involvement of autophagy in diseases of the heart or the brain, where the cells are fully differentiated and unlikely to be renewed or replaced, should be further explored. A lterations in organellophagy and lysosomal function should be evaluated in vivo with animal models or in patient derived tissues. Studying the roles of various organellophagy and lysosomal function in various disease processes will provide better understanding for the efficient manipulation of autophagy that may lead to novel therapeutic strategies. Targeting selective autophagy will allow the maintenance of homeostasis at each specific pathological location in a cell or organ. Determining the regulatory mechanism of each step in the autophagy–lysosome degradation pathway will be required in order to control the on- and off-target efficiency of each treatment at different stages.
I gratefully appreciate Dr. Qiangrong Liang and Derek Timm for their critical reading and revising of the manuscript. SK was supported by an American Heart Association (AHA) postdoctoral fellowship and by a Juvenile Diabetes Research Foundation (JDRF) advanced postdoctoral fellowship.
The author declares no conflict of interest.