Abstract
Human parainfluenza virus type 1 (hPIV1) does not form clear plaque by the conventional plaque formation assay because of slightly a cytopathic effects in many cell lines infected with hPIV1, thus making in virus titration, isolation and inhibitor evaluation difficult. We have succeeded in fluorescent histochemical visualization of sialidase activities of influenza A and B viruses, Newcastle disease virus and Sendai virus by using a novel fluorescent sialidase substrate, 2-(benzothiazol-2-yl)-4-bromophenyl 5-acetamido-3,5-dideoxy-α-D-glycero-D-galacto-2-nonulopyranosidonic acid (BTP3-Neu5Ac). In this study, we applied the BTP3-Neu5Ac assay for rapid detection of hPIV1 and hPIV type 3. The BTP3-Neu5Ac assay could histochemically visualize dot-blotted hPIVs on a membrane and hPIV-infected cells as local fluorescence under UV irradiation. We succeeded in distinct fluorescent visualization of hPIV1-infected cells in only 3 d using the BTP3-Neu5Ac assay. Due to there being no fixation, hPIV1 was isolated directly from fluorescent stained focus cells by the BTP3-Neu5Ac assay. Establishment of a sensitive, easy, and rapid fluorescent focus detection assay for hPIV, hPIV1 in particular will contribute greatly to progress in hPIV studies.
Human parainfluenza virus type 1 (hPIV1) and human parainfluenza virus type 3 (hPIV3) belong to genus Respirovirus, subfamily Paramyxovirinae, and family Paramyxoviridae. The hPIVs cause flu-like respiratory symptoms, including croup, pneumonia and bronchiolitis, mainly in infants younger than 5 years old and immunocompromised patients. Severe symptoms often lead to hospitalization and occasionally result in sudden death of newborns and infants. hPIVs, the majority of which are hPIV1 and hPIV3, are detected from approximately 20% of viral pneumonia cases in infants younger than 5 years of age.1,2) Although hPIVs are clinically important respiratory viruses for infants and immunocompromised patients, vaccines and medicines against hPIVs have not been developed.
Clinical samples and virus culture supernatants include some viruses and mutants. When viruses replicate in the presence of a certain inhibitor, inhibitor-resistant mutant viruses may appear in the culture supernatant. To separate a virus isolate from such samples, the plaque formation assay is frequently used. A virus infects a cell and produces progeny viruses that spread to infect neighboring cells. The population of infected cells shows a cytopathic effect (CPE), for example, cell death or syncytium formation. When virus-cultured cells are overlaid by a medium containing agarose, cell lysis induced by CPE is visualized as a plaque. A virus isolate can be picked up directly from a plaque, which is formed by an isolate. hPIV3 has been reported to form large plaques in human epidermoid cancer (HEp-2) cells, in which hPIV3 shows much syncytium formation.3) On the other hand, hPIV1 does not form plaques by the normal plaque formation method because of the weak CPE of hPIV1 in many cell lines.4) We established a plaque formation assay for hPIV1 by using neutral red in addition to a bovine serum albumin-free medium. Live cells incorporate neutral red via an endocytic pathway and their color becomes red. hPIV1-infected cells became weak and did not incorporate neutral red and thus remained white in color. By this method, visualization of hPIV1 plaques took 7 to 9 d.5) The plaque formation method is also frequently used for titration of virus infection and evaluation of virus inhibitors. The development of a faster and more sensitive plaque formation method would greatly contribute to progress in hPIV research fields including epidemiology, development of virus inhibitors, and characterization of virus isolates.
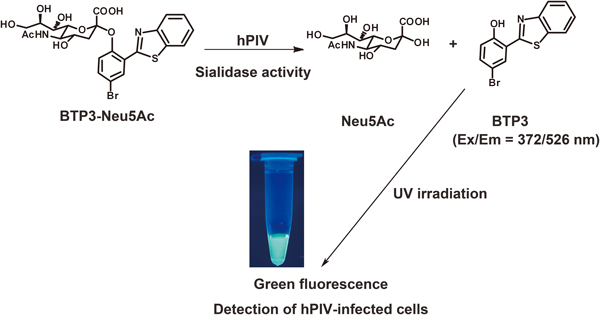
We have developed a novel fluorescent sialidase substrate, 2-(benzothiazol-2-yl)-4-bromophenyl 5-acetamido-3,5-dideoxy-α-D-glycero-D-galacto-2-nonulopyranosidonic acid (BTP3-Neu5Ac). The water-soluble non-fluorescent BTP3-Neu5Ac is cleaved to a water-insoluble fluorescent benzothiazolylphenol derivative (BTP3)6,7) and N-acetylneuraminic acid (Neu5Ac) by sialidase activity.8) BTP3 locally deposits on sialidase activity-existing sites, resulting in local and fluorescent visualization of sialidase activity in live cells (Fig. 1). For influenza A and B viruses, Sendai virus and Newcastle disease virus, we have also achieved local and fluorescent visualization of the virus and the virus-infected cells showing extremely high activity of viral sialidase within 10 min, with no need for cell fixation or specific anti-virus antibodies. BTP3-Neu5Ac also fluorescently visualized plaques of influenza A virus, from which a virus isolate could be obtained.9–11)
hPIVs have a hemagglutinin-neuraminidase (HN) glycoprotein that shows receptor binding ability to terminal sialic acids of glycochains and sialidase activity cleaving the terminal sialic acids from glycochains.12) Since BTP3-Neu5Ac can detect Sendai virus, one of the parainfluenza viruses (close to hPIV1),10) it is also predicted to be sensitive to hPIV sialidases (Fig. 1). We tried to establish a faster and more sensitive focus detection assay for hPIV1 by using BTP3-Neu5Ac. In the present study, we determined reaction conditions of hPIV1 and hPIV3 with BTP3-Neu5Ac, and we examined the utility of BTP3-Neu5Ac for fluorescent visualization of hPIV1-infected cells and focuses.
MATERIALS AND METHODS
ChemicalsBTP3-Neu5Ac and an hPIV1-sensitive sialidase inhibitor, a 5-acetamido-2,6-anhydro-3,5-dideoxy-D-glycero-non-2-enonic acid (Neu5Ac2en) derivative, 4-O-(2-thienyl-2-propynyl) Neu5Ac2en13) were synthesized by Dr. Tadamune Otsubo and Dr. Kiyoshi Ikeda at Hiroshima International University.
Cells and VirusesLLC-MK2 cells were maintained in Eagle’s minimum essential medium (MEM) supplemented with 5% fetal bovine serum (FBS). hPIV1 C35 strain and hPIV3 C243 strain were propagated in LLC-MK2 cells.
AntibodiesRabbit anti-hPIV1 polyclonal antibody was prepared by immunization of rabbits against hPIV1 C35 strain as described previously14) and purified on a Protein G column, HiTrapTM Protein G HP (CE Healthcare Biosciences Corp., Piscataway, NJ, U.S.A.). Horseradish peroxidase (HRP)-conjugated Protein A (Sigma-Aldrich, St. Louis, MO, U.S.A.) and phycoerythrin (PE)-conjugated goat anti-rabbit immunoglobulin G (IgG) (Santa Cruz Biotechnology, Inc., Dallas, TX, U.S.A.) were purchased.
Hemagglutination Unit DeterminationFifty microliters/well of 2-fold serial dilutions of hPIV1 and hPIV3 in phosphate-buffered saline (PBS; pH 7.2, 131 mM NaCl, 14 mM Na2HPO4, 1.5 mM KH2PO4, and 2.7 mM KCl) was poured into a U-bottom 96-well plate. PBS was used as a negative control without virus. Fifty microliters/well of 0.5% (v/v) guinea pig red blood cell suspension in PBS was added. A plate was incubated at 4°C for 2 h. The maximum dilution that showed hemagglutination was defined as the hemagglutination unit (HAU).
Fluorescent Visualization of Sialidase Activity of Dot-Blotted hPIV with BTP3-Neu5AcIn order to determine the optimal pH condition for hPIV sialidase activity in BTP3-Neu5Ac staining, dot-blotted hPIVs were reacted with BTP3-Neu5Ac at different pH values of 5 mM acetate buffer (pH 4.0, 4.5, 5.0, 5.5, and 6.0). We blotted 20 HAU of hPIV1 and 22 HAU of hPIV3 in 200 µL/dot on a polyvinylidene difluoride (PVDF) membrane and incubated virus blots in 100 µL/dot of 10 µM BTP3-Neu5Ac at pH 4.0, 4.5, 5.0, 5.5, and 6.0 for 10 min at room temperature. The membrane was washed with PBS. Fluorescent images of the membranes were obtained using Lumi Vision PRO HR (AISIN SEIKI Co., Ltd., Aichi, Japan) with a DR655 filter and a YA3 filter (Kenko Tokina Co., Ltd., Tokyo, Japan) under UV irradiation.
In order to evaluate the dependency of BTP3-Neu5Ac on hPIV sialidase activity, a PVDF membrane was dot-blotted with 20 to 2−11 HAU of hPIV1 and 23 to 2−8 HAU of hPIV3 in 200 µL/dot of PBS and incubated with 10 µM BTP3-Neu5Ac in 5 mM acetate buffer at pH 4.5 containing 1 mM CaCl2 for 10 min at room temperature. In addition, a PVDF membrane was dot-blotted with 24 to 2−6 HAU of hPIV1 in PBS at room temperature for 10 min. Fluorescent images of the membranes were obtained with Lumi Vision PRO HR under UV irradiation.
Fluorescent Visualization of hPIV-Infected Cells with BTP3-Neu5AcA confluent monolayer of LLC-MK2 cells on a 96-well plate was infected with the indicated focus-forming units (ffu) of hPIV1 and hPIV3 in 100 µL/well of serum-free medium (SFM), Hybridoma-SFM (Invitrogen Corp., Carlsbad, CA, U.S.A.), at 37°C for 48 h in 5% CO2. The cells were washed with PBS and reacted with 25 µM BTP3-Neu5Ac in 50 µL of SFM (adjusted to pH 4.5 with HCl) at 37°C for 5 min (hPIV1) or 15 min (hPIV3). The cells were observed with an IX71 fluorescent microscope (Olympus Co., Ltd., Tokyo, Japan) equipped with fluorescent filters (U-MWU2, DM400, BP336-385, BA420), under UV irradiation.
To confirm viral sialidase dependency of fluorescent staining with BTP3-Neu5Ac, hPIV1-infected cells were stained with 25 µM BTP3-Neu5Ac in 50 µL of SFM (adjusted to pH 4.5 with HCl) at 37°C for 5 min in the presence of 10 µM 4-O-(2-thienyl-2-propynyl) Neu5Ac2en. Inhibition of fluorescent staining with BTP3-Neu5Ac was observed with an IX71 fluorescent microscope.
Double Staining of hPIV1-Infected Cells with BTP3-Neu5Ac and Anti-hPIV1 AntibodyIn order to confirm that BTP3-Neu5Ac stained hPIV1-infected cells, infected cells were immunostained with a rabbit anti-hPIV1 polyclonal antibody. A confluent monolayer of LLC-MK2 cells on a 96-well plate was infected with the indicated ffu of hPIV1 in 100 µL/well of SFM at 37°C for 48 h in 5% CO2 and washed with 100 µL/well of PBS. Then the cells were fixed with 4% paraformaldehyde (Wako Pure Chemical Industries, Ltd., Osaka, Japan) at room temperature for 10 min and washed with PBS. The cells were treated with a rabbit anti-hPIV1 polyclonal antibody in PBS, followed by PE-conjugated goat anti-rabbit IgG at room temperature for 30 min each. Then the cells were incubated with 20 µM BTP3-Neu5Ac in 50 µL/well of 5 mM acetate buffer (pH 4.5) containing 1 mM CaCl2 at 37°C for 15 min. Immunostaining images of the infected cells were observed with an IX71 fluorescent microscope equipped with fluorescent filters (U-MWIG3, DM570, BP530–550, BA575IF).
Fluorescent Staining of hPIV1 Focuses with BTP3-Neu5AcThe hPIV1 plaque forming assay was performed using the protocol described previously with some modifications.5,15–17) Briefly, LLC-MK2 cells were seeded in a 6-well plate and cultured for 1 d until they became confluent. The cells were washed twice with 1 mL/well of PBS and infected with the indicated ffu of hPIV1 at room temperature for 1 h. Then the cells were washed with 1 mL/well of PBS and overlaid with 4 mL/well of the overlay medium, which contained 0.5% GTG Seakem Gold Agarose (Lonza Ltd., Basel, Switzerland) and 3 µg/mL acetylated trypsin (Sigma-Aldrich, St. Louis, MO, U.S.A.) in SFM. After the plate had been left at room temperature for 15 min to firm the overlay medium, the plates were incubated upside-down at 37°C. At 48 h post infection, 200 µM BTP3-Neu5Ac in 100 µL/well SFM was poured onto the overlay medium of the plate. The plates were incubated at 37°C overnight. Focuses were detected under UV irradiation at 365 nm.
Isolation of hPIV1 from Fluorescent FocusesWe examined whether hPIV1 could be isolated from fluorescent focuses stained with BTP3-Neu5Ac. A newly confluent monolayer of LLC-MK2 cells was washed with 1 mL/well of PBS and then 2 mL/well of SFM containing 3 µg/mL acetylated trypsin was added. The cells were infected by addition of respective suspensions in SFM obtained from three fluorescent focuses stained with BTP3-Neu5Ac. At 48 h post infection, the cells were washed with 1 mL/well of PBS and fixed with methanol at room temperature for 30 s and then washed twice with 1 mL/well of PBS. hPIV-infected cells were confirmed by immunostaining with anti-hPIV1 polyclonal antibody and HRP-conjugated Protein A using N,N-diethyl-p-phenylenediamine sulfate and 4-chloro-1-naphtol as described previously.18) To immunostain hPIV1 focuses, the cells in a plate were fixed by pouring 3 mL/well of ethanol–acetic acid (5 : 1, v/v) onto the overlay medium at 4°C overnight. The overlay medium was removed. The cells were washed twice with PBS and fixed with 800 µL/well of methanol for 30 s. Then the cells were washed with PBS and incubated with rabbit anti-hPIV1 antibody at room temperature for 30 min, followed by HRP-conjugated Protein A at room temperature for 30 min. The infected cells were washed with PBS and stained with N,N-diethyl-p-phenylenediamine sulfate and 4-chloro-1-naphtol as described previously.18)
RESULTS
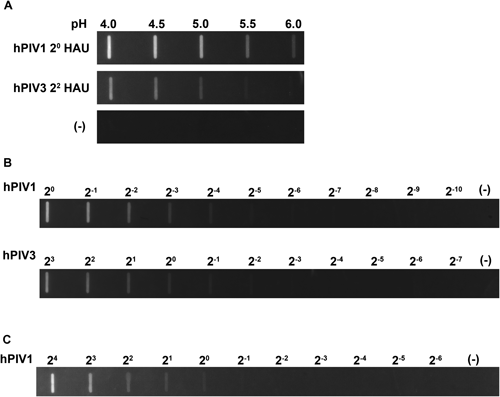
Fluorescent Visualization of hPIV1 Dot-Blotted on a Membrane with BTP3-Neu5AcSialidase activities of hPIV1 and hPIV3 have been reported to be the most reactive at acidic pH, optimal pH 4.0–4.5 for hPIV119) and optimal pH 4.7 for hPIV3.20) In addition, we succeeded in fluorescent visualization of Sendai virus sialidase under a BTP3-Neu5Ac reaction at pH 4.5.10) It is estimated that acidic conditions are suitable for hPIV sialidases in BTP3-Neu5Ac reaction. We reacted dot-blotted hPIV1 and hPIV3 on a PVDF membrane with BTP3-Neu5Ac in acetate buffers with different pH values (pH 4.0, 4.5, 5.0, 5.5, and 6.0). After incubation with 10 µM BTP3-Neu5Ac at room temperature for 10 min, both hPIV1 and hPIV3 showed the highest fluorescent intensity at pH 4.0 and 4.5 (Fig. 2A). We planned to apply pH 4.5 for more sensitive detection of dot-blotted hPIVs and hPIV-infected cells with BTP3-Neu5Ac.
We examined the dependency of BTP3-Neu5Ac reaction on amounts of hPIV1 and hPIV3, meaning that the fluorescence is dependent on the virus sialidase. hPIV1 and hPIV3 were 2-fold serially diluted and blotted onto a PVDF membrane. The membrane was incubated with 10 µM BTP3-Neu5Ac in 5 mM acetate buffer (pH 4.5) containing 1 mM CaCl2 at room temperature for 10 min. Ca2+ ion was previously reported to boost hPIV1 sialidase activity.19,21,22) Both dot-blotted hPIV1 and hPIV3 were fluorescently visualized in a manner dependent on the amount of hPIV under UV irradiation. At least 2−4 HAU for hPIV1 and 2° HAU for hPIV3 were distinctly detected by incubation of 10 µM BTP3-Neu5Ac for 10 min (Fig. 2B). We confirmed that BTP3-Neu5Ac is a sensitive and specific substrate of hPIV1 and hPIV3 sialidases.
hPIV1 sialidase was faintly reactive to BTP3-Neu5Ac even at pH 6.0 (Fig. 2A). An acidic condition was more preferable for detection of hPIV sialidases using BTP3-Neu5Ac. However, fluorescent visualization at neutral pH would be more suitable for hPIV proliferation and isolation directly from fluorescent cells because of less damage of cells and viruses. Dot-blotted hPIV1 on a PVDF membrane was incubated with 10 µM BTP3-Neu5Ac in neutral pH of PBS at room temperature for 10 min. Even in a neutral condition, BTP3-Neu5Ac was able to detect hPIV1 until at least 2° HAU, although the sensitivity was lower than that under an acidic condition (Fig. 2C).
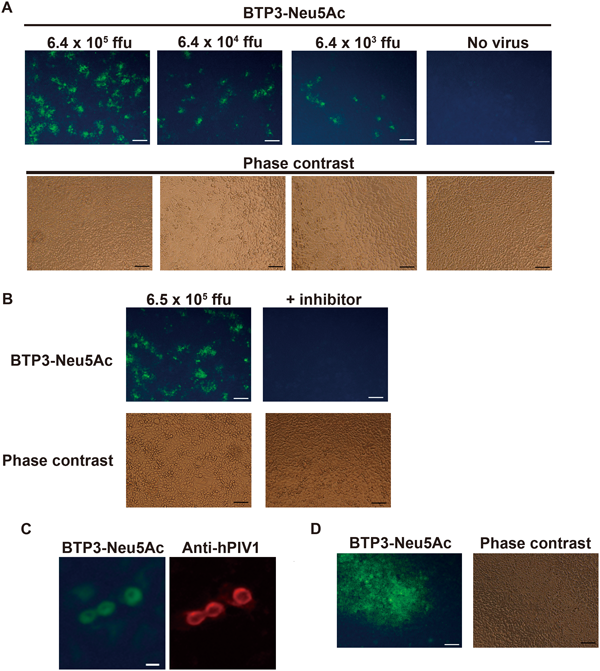
Fluorescent Visualization of hPIV-Infected Cells with BTP3-Neu5AcHN glycoprotein is abundantly expressed on the surface membrane of hPIV-infected cells, meaning that the hPIV-infected cells have high virus sialidase activity. We expected that BTP3-Neu5Ac could specifically and easily visualize hPIV-infected cells in a short time, with no need for cell fixation or specific anti-virus antibody. Since the sensitivity of BTP3-Neu5Ac for hPIV1 is higher than that for hPIV3, we used hPIV1 in almost all experiments for fluorescent visualization of infected cells. LLC-MK2 cells, which are usually used in experiments on hPIV infection and proliferation, were infected with hPIV1 for 48 h. The infected cells were treated with 25 µM BTP3-Neu5Ac in SFM (pH 4.5) at 37°C for 5 min. Green fluorescence-stained cells increased in a manner dependent on the amount of hPIV1 infection. There was no fluorescence in non-infected cells (Fig. 3A). In order to confirm that the fluorescence is specifically derived from virus sialidase activity and not from endogenous mammalian sialidase activity, hPIV1-infected cells were reacted with BTP3-Neu5Ac in the presence of an hPIV1 sialidase-sensitive inhibitor, 4-O-(2-thienyl-2-propynyl) Neu5Ac2en, which was developed in our previous study.13) The sialidase inhibitor completely inhibited fluorescent visualization of the hPIV1-infected cells with BTP3-Neu5Ac (Fig. 3B). It was confirmed that BTP3-Neu5Ac visualized cells by hPIV1 sialidase activity. Moreover, hPIV1-infected cells were double stained with both anti-hPIV1 polyclonal antibody and BTP3-Neu5Ac. At 48 h post infection, the infected cells were fixed and incubated with anti-hPIV1 antibody, followed by a secondary antibody. Then the cells were incubated with BTP3-Neu5Ac at 37°C for 15 min. hPIV1 antigen-positive cells were almost merged with BTP3-Neu5Ac staining (Fig. 3C). Taken together, we concluded that BTP3-Neu5Ac distinctly visualized hPIV1-infected cells. In addition to the results for hPIV1, we also succeeded in detection of hPIV3-infected cells at 48 post infection by incubation with 25 µM BTP3-Neu5Ac at 37°C for 15 min (Fig. 3D).
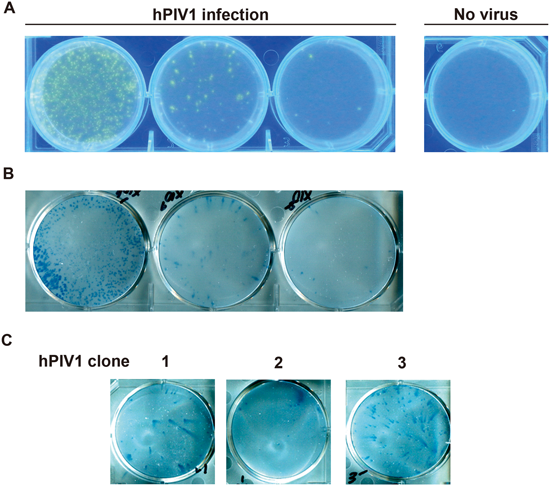
Fluorescent Visualization of hPIV1 Focuses by Using BTP3-Neu5AcAn infected cell releases many progeny viruses, which spread to infect neighboring cells. For the conventional plaque forming assay, the overlay medium containing agarose on infected cells restricts the spread of progeny virus infection to neighboring cells. Such infections result in a CPE (cell death and syncytium formation, etc.) of infected cell populations, which can be detected as visible plaques. However, since hPIV1 is one of the viruses showing little CPE, it is extremely difficult to apply to the conventional plaque formation method. We tried to establish fluorescent visualization of hPIV1 focuses by using BTP3-Neu5Ac. In accordance with the conventional plaque forming assay, LLC-MK2 cells were infected with hPIV1 and cultured on an overlay medium containing 0.5% agarose at 37°C for 48 h. Two hundred micromolars of BTP3-Neu5Ac in SFM (neutral pH) was poured onto the overlay medium (20 µM BTP3-Neu5Ac as a final concentration) and incubated at 37°C overnight. Fluorescent focuses increased in a manner dependent on the amount of hPIV1 infection under UV irradiation at 365 nm (less gene damage due to high wave length) (Fig. 4A). We succeeded in distinct fluorescent visualization of focuses without cell fixation and a specific anti-virus antibody. Focuses were also confirmed by immunostaining with anti-hPIV1 antibody, indicating that BTP3-Neu5Ac enables more distinct visualization of focuses than does immunostaining (Fig. 4B). We showed that our focus formation assay is useful for virus isolation. Three focuses were selected and independently cultured in newly confluent LLC-MK2 cells for 48 h. The infected cells were immunostained with anti-hPIV1 antibody. For all of the three selected fluorescent focuses, we detected infection and proliferation of isolated hPIV1 clones in LLC-MK2 cells by immunostaining (Fig. 4C). We succeeded in detection of hPIV1 focuses (in fact, infected cell populations) in only 3 d by using BTP3-Neu5Ac and also succeeded in hPIV1 isolation directly from fluorescent focuses without cell fixation.
Discussion and Conclusion
In the present study, we established a sensitive, rapid, and easy method for fluorescent visualization of hPIV1- and hPIV3-infected cells by using BTP3-Neu5Ac. We also demonstrated the utility of hPIV1 isolation directly from fluorescent focuses of hPIV1. The BTP3-Neu5Ac assay does not require cell fixation, a specific anti-virus antibody or secondary antibody, and it enables easy and rapid detection (in 5–20 min). Regarding the time required until visualization of hPIV1 focuses, our method (in 3 d) is much more rapid than the previous method using neutral red (in 7–9 d).5)
There is a concern that fluorescence after BTP3-Neu5Ac reaction may be derived from endogenous mammalian sialidase activity other than virus sialidase activity. We therefore confirmed an inhibitory effect of an hPIV1 sialidase-specific inhibitor and co-localization by immunostaining with anti-hPIV1 antibody and by BTP3-Neu5Ac staining. Fluorescence after BTP3-Neu5Ac reaction increased in a manner dependent on the amount of dot-blotted hPIV and hPIV1 infection. Of course, there was no fluorescence in non-infected cells. We also confirmed that HN-expressed cells, which were transfected with each HN gene from hPIV1 or hPIV3, were stained by the BTP3-Neu5Ac assay (data not shown), further suggesting that the fluorescence resulted from hPIV sialidase activity. In our previous study, fluorescent staining of mammalian sialidase activity in the rat brain needed incubation of 1 mM BTP3-Neu5Ac for at least 1 h.8) Incubation of 10–25 µM BTP3-Neu5Ac for only 5–15 min is sufficient for staining of hPIV and hPIV-infected cells, meaning much lower concentrations of BTP3-Neu5Ac and much shorter reaction time than those for staining of mammalian sialdiase activity. These results strongly suggest that the fluorescence of hPIV-infected cells after BTP3-Neu5Ac reaction is due to the virus sialidase activity.
The fluorescent compound BTP3 has an acid resistance for its fluorescence property,6) which enabled sialidase reaction under an acidic condition in the present study. BTP3 also has a large stokes shift, which is an excitation of UV (372 nm at maximum wave length) and an emission of green fluorescence (526 nm at maximum wave length). 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI), fluorescein isothiocyanate (FITC), and tetramethylrhodamine (TRITC) are representative fluorescent compounds frequently used for fluorescent staining. DAPI, FITC, and TRITC emit blue, green, and red fluorescence by excitations of UV, blue, and green lasers, respectively. Since BTP3 can distinctly discriminate these representative fluorescent compounds by their fluorescent properties, BTP3-Neu5Ac enables co-staining together with these compounds. In fact, we performed double staining of hPIV1-infected cells with BTP3-Neu5Ac and PE. BTP3-Neu5Ac is thus useful for multicolor staining.
Detection of virus-infected cells and focuses is frequently used in various virological studies and hygiene surveys, including confirmation of virus proliferation and determination of virus infectious titers as focus-forming units. This focus formation assay is also useful for screening of reagents inhibiting virus infection and proliferation, discovery of antiviral reagent-resistant viruses, and investigation of genetic mutations involved in viral infectivity, replicability, and pathogenesis. An immunostaining method is not appropriate for isolating viruses because the viruses are inactivated by cell fixation and antivirus antibody treatment. hPIV1 is extremely difficult to apply to the conventional plaque forming assay because of the weak CPE of hPIV1-infected cells. Visualization of the virus with the BTP3-Neu5Ac assay will greatly contribute to progress in research on hPIVs.
Acknowledgments
We thank Dr. Allen Portner (St. Jude Children’s Research Hospital) for providing virus strains. This work was in part supported by a Grant from Nakatani Foundation for Advancement of Measuring Technologies in Biomedical Engineering, the Kurata Memorial Hitachi Science and Technology Foundation, the Futaba Electronics Memorial Foundation, Tokai Foundation for Technology, Takahashi Industrial and Economic Research Foundation, Izumi Science and Technology Foundation, The Shimabara Science Promotion Foundation, Research Foundation for the Electrotechnology of Chubu, Tokyo Biomarker Innovation Research Association (TOBIRA), Japan Foundation for Aging and Health, The Pharmacological Research Foundation, Tokyo, University of Shizuoka, and JSPS KAKENHI Grant Numbers Challenging Exploratory Research 26670064 and Young Scientists (A) 15H05644. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Henrickson KJ. Parainfluenza viruses. Clin. Microbiol. Rev., 16, 242–264 (2003).
- 2) Sinaniotis CA. Viral pneumoniae in children: incidence and aetiology. Paediatr. Respir. Rev., 5 (Suppl. A), S197–S200 (2004).
- 3) McKimm-Breschkin JL. A simplified plaque assay for respiratory syncytial virus-direct visualization of plaques without immunostaining. J. Virol. Methods, 120, 113–117 (2004).
- 4) Tao T, Durbin AP, Whitehead SS, Davoodi F, Collins PL, Murphy BR. Recovery of a fully viable chimeric human parainfluenza virus (PIV) type 3 in which the hemagglutinin-neuraminidase and fusion glycoproteins have been replaced by those of PIV type 1. J. Virol., 72, 2955–2961 (1998).
- 5) Fukushima K, Takahashi T, Takaguchi M, Ueyama H, Ito S, Kurebayashi Y, Kawanishi T, McKimm-Breschkin JL, Takimoto T, Minami A, Suzuki T. Plaque formation assay for human parainfluenza virus type 1. Biol. Pharm. Bull., 34, 996–1000 (2011).
- 6) Otsubo T, Minami A, Fujii H, Taguchi R, Takahashi T, Suzuki T, Teraoka F, Ikeda K. 2-(Benzothiazol-2-yl)-phenyl-β-D-galactopyranoside derivatives as fluorescent pigment dyeing substrates and their application for the assay of β-D-galactosidase activities. Bioorg. Med. Chem. Lett., 23, 2245–2249 (2013).
- 7) Takahashi T, Otsubo T, Ikeda K, Minami A, Suzuki T. Histochemical imaging of alkaline phosphatase using a novel fluorescent substrate. Biol. Pharm. Bull., 37, 1668–1673 (2014).
- 8) Minami A, Otsubo T, Ieno D, Ikeda K, Kanazawa H, Shimizu K, Ohata K, Yokochi T, Horii Y, Fukumoto H, Taguchi R, Takahashi T, Oku N, Suzuki T. Visualization of sialidase activity in mammalian tissues and cancer detection with a novel fluorescent sialidase substrate. PLoS ONE, 9, e81941 (2014).
- 9) Kurebayashi Y, Takahashi T, Otsubo T, Ikeda K, Takahashi S, Takano M, Agarikuchi T, Sato T, Matsuda Y, Minami A, Kanazawa H, Uchida Y, Saito T, Kawaoka Y, Yamada T, Kawamori F, Thomson R, von Itzstein M, Suzuki T. Imaging of influenza virus sialidase activity in living cells. Sci. Rep., 4, 4877 (2014).
- 10) Takano M, Takahashi T, Agarikuchi T, Kurebayashi Y, Minami A, Otsubo T, Ikeda K, Kanazawa H, Suzuki T. Histochemical fluorescent staining of Sendai virus-infected cells with a novel sialidase substrate. Virology, 464–465, 206–212 (2014).
- 11) Takahashi T, Takano M, Agarikuchi T, Kurebayashi Y, Minami A, Otsubo T, Ikeda K, Suzuki T. A novel method for detection of Newcastle disease virus with a fluorescent sialidase substrate. J. Virol. Methods, 209, 136–142 (2014).
- 12) Fukushima K, Takahashi T, Ito S, Takaguchi M, Takano M, Kurebayashi Y, Oishi K, Minami A, Kato T, Park EY, Nishimura H, Takimoto T, Suzuki T. Terminal sialic acid linkages determine different cell infectivities of human parainfluenza virus type 1 and type 3. Virology, 464–465, 424–431 (2014).
- 13) Sato K, Ikeda K, Suzuki T, Aoyama S, Maki N, Suzuki S, Sato M. Synthesis of 4-O-[3-(aryl)prop-2-ynyl]-Neu5Ac2en and its 4-epi-analogs modified at C-4 by Sonogashira coupling reaction. Tetrahedron, 63, 7571–7581 (2007).
- 14) Suzuki T, Ikeda K, Koyama N, Hosokawa C, Kogure T, Takahashi T, Jwa Hidari KI, Miyamoto D, Tanaka K, Suzuki Y. Inhibition of human parainfluenza virus type 1 sialidase by analogs of 2-deoxy-2,3-didehydro-N-acetylneuraminic acid. Glycoconj. J., 18, 331–337 (2001).
- 15) Shibuta H, Akami M, Matumoto M. Plaque formation by sendai virus of parainfluenza virus group, type 1 on monkey, calf kidney and chick embryo cell monolayers. Jpn. J. Microbiol., 15, 175–183 (1971).
- 16) Sugita K, Maru M, Sato KA. Sensitive plaque assay for Sendai virus in an established line of monkey kidney cells. Jpn. J. Microbiol., 18, 262–264 (1974).
- 17) Scheid A, Choppin PW. Protease activation mutants of Sendai virus. Activation of biological properties by specific proteases. Virology, 69, 265–277 (1976).
- 18) Takahashi T, Murakami K, Nagakura M, Kishita H, Watanabe S, Honke K, Ogura K, Tai T, Kawasaki K, Miyamoto D, Hidari KI, Guo CT, Suzuki Y, Suzuki T. Sulfatide is required for efficient replication of influenza A virus. J. Virol., 82, 5940–5950 (2008).
- 19) Wang ZM, Tong LL, Grant D, Cihlar T. Expression and characterization of soluble human parainfluenza virus type 1 hemagglutinin-neuraminidase glycoprotein. J. Virol. Methods, 98, 53–61 (2001).
- 20) Moscona A, Peluso RW. Relative affinity of the human parainfluenza virus type 3 hemagglutinin-neuraminidase for sialic acid correlates with virus-induced fusion activity. J. Virol., 67, 6463–6468 (1993).
- 21) Merz DC, Prehm P, Scheid A, Choppin PW. Inhibition of the neuraminidase of paramyxoviruses by halide ions: a possible means of modulating the two activities of the HN protein. Virology, 112, 296–305 (1981).
- 22) Tappert MM, Smith DF, Air GM. Fixation of oligosaccharides to a surface may increase the susceptibility to human parainfluenza virus 1, 2, or 3 hemagglutinin-neuraminidase. J. Virol., 85, 12146–12159 (2011).