Notes
Regulation of Histone Deacetylase 6 Activity via S-Nitrosylation
2015 Volume 38 Issue 9 Pages 1434-1437
Details
2015 Volume 38 Issue 9 Pages 1434-1437
Nitric oxide (NO) is a gaseous regulatory factor produced by NO synthases (NOS) and it plays several critical roles via S-nitrosylation of protein cysteine residues. Histone deacetylase (HDAC) functions in the maintenance/balance of chromatin acetylation and contributes to transcriptional supression. It has been reported that S-nitrosylation of HDAC2 is involved in the regulation of deacetylase activity. However, it remains unknown whether other subtypes of the HDAC family are S-nitrosylated. In the present study, we found that HDAC6 is a target of NO. A biotin-switch assay revealed that endogenous HDAC6 is S-nitrosylated by both NO donors and NO derived from the inducible type of NOS in cells treated with cytokines. NO led to suppressed deacetylase activity in vitro and increased acetylated α-tubulin, a major substrate for HDAC6, in A549 cells. These findings suggest that S-nitrosylation of HDAC6 plays a pivotal role in the regulation of protein acetylation.
Nitric oxide (NO) is a highly diffusible molecule generated by a family of NO synthases (NOSs) that convert L-arginine to L-citrulline using oxygen and nicotine adenine dinucleotide phosphate (NADPH).1) Under physiological conditions, NO is produced at low levels in response to endogenous stimuli, such as acetylcholine or bradykinin, and acts as a critical messenger of intracellular signaling pathways. NO also has a variety of physiological functions, such as neurotransmission, inflammatory reaction, vasodilation, and stimulates both angiogenesis and cell proliferation.2,3) However, excess amounts of NO can result in pathophysiological conditions such as neurodegenerative diseases, hypertension, and arterial sclerosis.4)
Protein S-nitrosylation is an NO-dependent reversible post-translational modification of cysteine that regulates both protein structure and function in bacteria, plants, and mammals.5,6) An activated NO moiety is covalently bound to the selective free thiol group on cysteine, generating an S-nitrosothiol. Over the past couple of decades, advanced proteomic approaches have led to the identification of more than a thousand S-nitrosylated (SNO-) proteins with diverse cellular functions. Previously, we demonstrated that protein disulfide isomerase (PDI) and phosphatase and tensin homolog deleted from chromosome 10 (PTEN) are targets of NO and regulate its enzymatic activity via S-nitrosylation.7,8)
S-Nitrosylation of histone deacetylase 2 (HDAC2) regulates its function and induces epigenetic changes in neuronal cells and hepatocytes, although it remains unclear as to whether NO regulates other HDACs via S-nitrosylation.9,10) On the other hand, HDAC6 is localized in the cytoplasm, not the nucleus, and plays a key role in regulating cell motility and signaling through deacetylation of substrate proteins such as α-tubulin, and heat shock protein 90 (HSP90) in human lung carcinoma A549 cells.11–14) Here, we found that HDAC6 is S-nitrosylated in A549 cells expressing inducible NOS (iNOS). Interestingly, treatment with NO resulted in the attenuation of HDAC6 deacetylase activity in A549 cells, implicating that endogenous S-nitrosylation of HDAC6 in the control of protein acetylation.
The human lung cancer cell line A549 was purchased from the RIKEN BRC (Saitama, Japan). N-[6-(Biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide (HPDP-biotin) was purchased from Pierce Chemical Co. (Rockford, IL, U.S.A.). Human recombinant tumor necrosis factor (TNF)-α, interleukin (IL)-1β and interferon (IFN)-γ were purchased from PEPRO TECH, Inc. (Rocky Hill, NJ, U.S.A.). NG-Nitro-L-arginine methyl ester (L-NAME) was obtained from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Tubastatin A was purchased from Selleck Chem. (Huston, TX, U.S.A.). Anti-HDAC6 and anti-iNOS antibodies were obtained from Cell Signaling Technology (Beverly, MA, U.S.A.) and EMD Millipore (Billerica, MA, U.S.A.), respectively. Anti-acetyl-α-tubulin and anti-α-tubulin were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.).
Cell CultureHuman lung adenocarcinoma A549 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin. At approximately 70% confluency, cultures were switched to serum-free medium for 24 h and stimulated with cytokines (10 ng/mL TNF-α+500 Units/mL IL-1β+100 Units/mL IFN-γ) in serum-free medium from 6 to 24 h.15) L-NAME was used as an NOS inhibitor and pre-incubated for 1 h prior to cytokine stimulation.
Western Blot AnalysisA549 cells were treated as indicated, then harvested, washed with phosphate-buffered saline (PBS), and lysed in ice-cold lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM ethylenediamine N,N,N′,N′-tetraacetic acid (EDTA), and 1% NP-40 containing a protease inhibitor cocktail) for 10 min. After quantification by the Bradford assay, proteins were boiled in Laemmli buffer for 5 min and separated by electrophoresis. Following sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE), proteins were transferred to polyvinylidene difluoride (PVDF) membranes and blocked with 5% non-fat dry milk or bovine serum albumin (BSA) in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 30 min at room temperature. Next, membranes were incubated with anti-HDAC6 (1 : 2000), anti-iNOS (1 : 10000), anti-acetyl-α-tubulin (1 : 2000), or anti-α-tubulin (1 : 5000) antibodies followed by anti-horseradish peroxidase (HRP)-linked secondary antibodies. The antibody-reactive bands were visualized by enhanced chemiluminescence (ECL). Blots were quantified using Image J software and relative ratios were calculated.
Biotin-Switch Assay for SNO-HDAC6Cell lysates were prepared in ice-cold HENT buffer (250 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid-sodium hydroxide (HEPES-NaOH), pH 7.5, 1 mM EDTA, 0.1 mM neocuproine, and 1% TritonX-100). Protein (0.8 mg) was used for the biotin-switch assay. Blocking buffer (2.5% SDS and 50 mM methylmethane thiosulfonate [MMTS] in HEN buffer) was mixed with the samples and incubated for 20 min at 50°C to block free thiol groups. After removing excess MMTS by acetone precipitation, nitrosothiols were reduced to thiols with 25 mM sodium ascorbate. Then, the newly formed thiols were then linked to sulfhydryl-specific biotinylation reagent, HPDP-biotin. Biotinylated proteins were pulled down using streptavidin agarose beads, and Western blotting was performed to determine the amount of HDAC6 remaining in the samples.
Deacetylase Activity of HDAC6The measurement of an in vitro deacetylase activity of HDAC6 was performed as described.16) Human recombinant HDAC6 was mixed with HD buffer (20 mM Tris–HCl, pH 8.0, 150 mM NaCl, 10% glycerol) and incubated with or without the indicated concentrations of S-nitrosocysteine (SNOC) for 1 h at room temperature in 96-well black half-area microplates (Greiner Bio-One). HDAC6-specific inhibitor, tubastatin A, was used as a positive control. Tubastatin A was incubated with recombinant HDAC6 for 10 min at room temperature. Next, 100 µM substrate peptides (acetylated peptidyl methylcoumarinamide (MCA)) were added and incubated for an additional 30 min at 37°C. At this time, acetylated peptidyl MCA was converted into peptidyl MCA by the deacetylase activity of HDAC6. Peptidyl MCA was decomposed to its fluorescent form, 7-amino-4-methylcoumarin (AMC), by trypsin treatment for 15 min at 37°C. Deacetylase activity was determined by measuring fluorescence intensity at excitation wavelength 370 nm and emission wavelength 460 nm.
Statistical AnalysisAll experiments were repeated independently at least three times. Results are presented as the mean±standard deviation (S.D.). Statistical comparisons were performed using Student’s t-tests or one-way ANOVA post hoc Bonferroni’s test using Graphpad Prism 5 (Graphpad Software, La Jolla, CA, U.S.A.). A p value <0.05 was considered to indicate statistical significance.
Since HDAC6 is involved in the aggresome formation/clearance, regulation of HSP90 activity, and cell survival in A549 cells,11–14) we therefore used this cell line to assess the effect of S-nitrosylation on HDAC6 activity. To address whether HDAC6 is S-nitrosylated by NO, we initially exposed cell lysates prepared from A549 cells to physiological NO donors, SNOC and S-nitrosoglutathione (GSNO). Then, a biotin-switch assay was performed to detect S-nitrosylated HDAC6 (SNO-HDAC6) as described previously.17) As shown in Fig. 1A, we found that both NO donors increase the level of SNO-HDAC6 in a concentration-dependent manner. Then we investigated whether SNO-HDAC6 formation is detectable in cells. Exposure to SNOC resulted in elevated SNO-HDAC6 levels in a dose-dependent fashion (Fig. 1B). To determine whether HDAC6 is S-nitrosylated by endogenously produced NO, A549 cells were stimulated with a mixture of inflammatory cytokines (TNF-α, IL-1β and IFN-γ) to induce iNOS expression.15,18) It is widely accepted that incubation of A549 cells with cytokines induces iNOS expression.15,18) The accumulation of nitrite, a metabolic product of NO, was measured in culture medium using the Griess assay. Treatment with cytokines resulted in the significant SNO-HDAC6 formation (Fig. 1C, upper panel). Time-dependent accumulation of nitrite was significantly detected following stimulation with cytokines in A549 cells (Fig. 1C, lower panel). These data indicate that HDAC6 is a possible target of NO in cells.
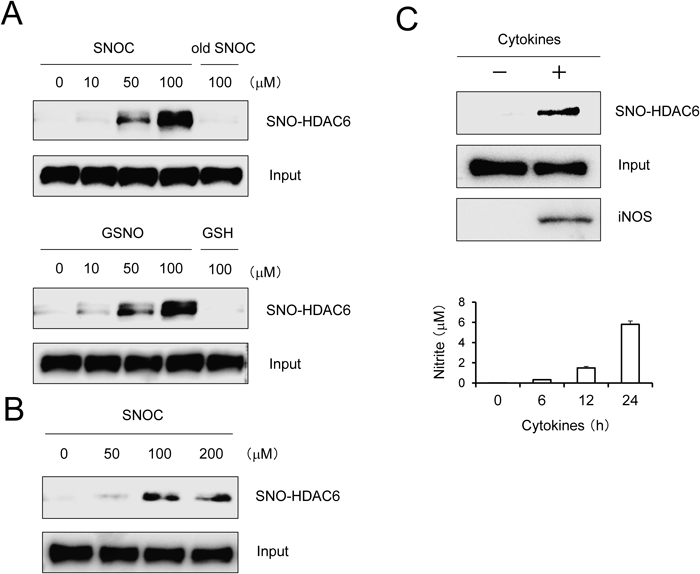
(A) Cell lysates from human A549 cells were incubated with SNOC or GSNO at room temperature for 30 min. Control samples were subjected to decayed (old) SNOC or GSH (reduced form glutathione). SNO-HDAC6 was detected using the biotin-switch assay. Total HDAC6 (Input) in cell lysates was detected by Western blotting. (B) A549 cells were incubated with SNOC at 37°C for 1 h. SNO-HDAC6 was detected using biotin-switch assay. (C) Endogenous iNOS induction by cytokines increased SNO-HDAC6 formation. A549 cells were stimulated with cytokines (TNF-α, IL-1β and IFN-γ) for 24 h at 37°C. SNO-HDAC6 was detected using the biotin-switch assay (upper panel). Nitrite accumulation was measured by the Griess assay (lower panel).
We next attempted to determine whether NO influences HDAC6 enzymatic activity. In vitro assays showed that tubastatin A, an HDAC6 inhibitor, strongly attenuated the deacetylase activity of human recombinant HDAC6.19,20) On the other hand, SNOC treatment significantly attenuated the activity, indicating that NO directly inhibits its enzymatic activity (Fig. 2A). Furthermore, we examined the effects of NO on HDAC6 activity in A549 cells. The level of acetylated α-tubulin was significantly elevated upon treatment with cytokines (Figs. 2B, C). This increment was strongly suppressed by challenge with L-NAME, an NOS inhibitor (Fig. 2D).
HDAC6 is localized in the cytoplasm and mainly regulates the acetylation of proteins in mammalian cells while participating in protein trafficking, degradation, cell shape, migration, aggresome formation/clearance, and cell viabillity, in response to misfolded protein stress.14) The various roles of HDAC6 indicate that its enzymatic activity may be strictly controlled, whereas the modulation of HDAC6 activity by post-translational modification remains unclear. Recently, it was reported that the phosphorylation of HDAC6 is involved in the regulation of deacetylase activity.21) On the other hand, it has yet to be determined whether HDAC6 activity is influenced by redox state (e.g., S-nitrosylation). HDAC6 plays a crucial role in promoting aggresome formation of misfolded/ubiquitinated proteins that are found in pathological features common to neurodegenerative diseases, including Parkinson’s, Alzheimer’s, and Huntington’s.14,22) Misfolded proteins are generally degraded by the proteasome to avoid a pathological condition or transported to the aggresome with the cargo onto the dynein motor complex via HDAC6. This event is believed to participate in the reduction of intracellular severe stress evoked by an accumulation of misfolded proteins. In the present study, we successfully showed for the first time that HDAC6 could be a substrate of NO (Fig. 1). Interestingly, S-nitrosylation of HDAC6 resulted in the attenuation of its deacetylase activity and consequently elevated the acetylated level of α-tubulin (Fig. 2). Here, we conclude that HDAC6 is regulated by redox state, especially S-nitrosylation. Excess amounts of NO may induce pathophysiological conditions such as neuronal cell death and related neurodegenerative disorders with an accumulation of misfolded proteins.7) Hence, the accumulation of misfolded proteins and subsequent cell death may be partly dependent on HDAC6 inactivation via S-nitrosylation.
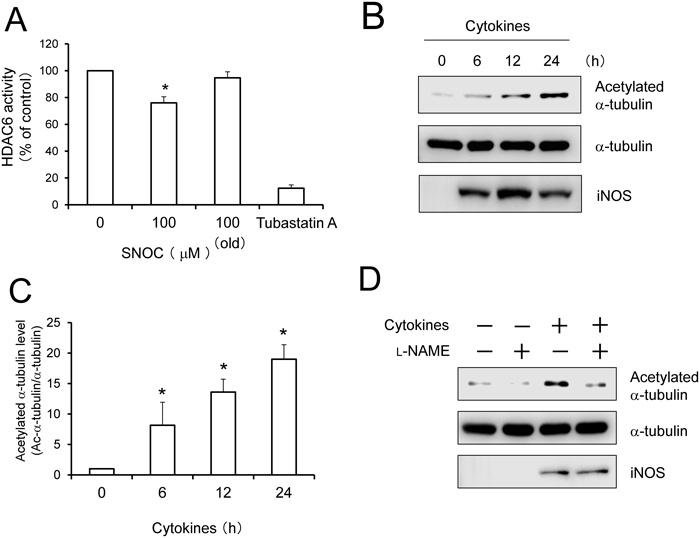
(A) Attenuation of HDAC6 enzymatic activity following treatment with SNOC in vitro. Human recombinant HDAC6 was pre-incubated with the substrate and SNOC (100 µM each) for 1 h and then incubated with trypsin. HDAC6 deacetylase activity was determined by measuring fluorescence intensity. Tubastatin A (1 µM) was pretreated for 1 h. Values are means±S.D., n=3; * p<0.05 by ANOVA. (B) Augmentation of acetylated α-tubulin levels by treatment with cytokines in A549 cells. Cells were stimulated with cytokines at 37°C for the indicated times. (C) The level of α-tubulin acetylation was quantified and normalized to the 0 h cytokines stimulation of the control group. Values represent the mean±S.D., n=3 experiments; * p<0.05 by ANOVA. (D) Effect of NOS inhibition on cytokine-induced acetylated α-tubulin. L-NAME (1 mM) was pretreated with cultured medium for 1 h prior to cytokine stimulation.
We thank Yoko Okamoto for technical assistance. This work was supported in part by the Japan Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan Grants-in-Aid for Scientific Research (B) 15H04649, Challenging Exploratory Research 15K14952; Takeda Science Foundation, the Uehara Memorial Foundation, and the Smoking Research Foundation.
The authors declare no conflict of interest.