Regular Articles
Effects of Glycerophospholipids on Ceramide Kinase Activity: Cardiolipin-Affected Cellular Formation of Ceramide-1-phosphate
2016 Volume 39 Issue 10 Pages 1708-1717
Details
2016 Volume 39 Issue 10 Pages 1708-1717
Ceramide kinase (CerK) and ceramide-1-phosphate (C1P) are involved in various cellular functions, while regulation of the enzyme activity has not been well elucidated. We herein investigated the effects of several glycerophospholipids on human recombinant CerK activity with CaCl2 and MgCl2 by measuring the formation of fluorescent labeled C1P in vitro. CerK activities were 44.1±11.4 (pmol/µg/min) with vehicle, 137±29 with 2 mM CaCl2, and 144±32 with 2 mM MgCl2 in the glycerol/albumin buffer. The addition of glycerophospholipids such as phosphatidylcholine, phosphatidylinositol (PI), PI 4,5-bisphosphate (PI(4,5)P2), and phosphatidic acid had no effect on CerK activity with CaCl2, although PI(4,5)P2 and phosphatidic acid bound to CerK in the lipid–protein overlay assay. The addition of cardiolipin (diphosphatidylglycerol) at concentrations up to 0.1 µM increased, whereas those more than 1 µM decreased CerK activity with CaCl2/MgCl2. In the lipid–protein overlay assay, cardiolipin bound to CerK and CerK lacking pleckstrin homology (PH) domain, but not PH domain of CerK, in CaCl2-independent manner. Cardiolipin also bound to CerK in the multilamellar vesicle binding assay. A deviation from the normal range of cellular cardiolipin, both the decrease by phospholipase D6 expression and increase by an exogenous addition of the lipid, negatively regulated C1P formation in intact HepG2 cells. Our results revealed that cardiolipin bound to CerK and regulated the formation of C1P in vitro and in cells.
Ceramide kinase (CerK) produces ceramide-1-phosphate (C1P) through the phosphorylation of ceramide, and the cloning and functional characterization of this enzyme were successfully achieved by Drs. Kohama and Spiegel’s group.1) C1P and ceramide both act as intracellular and extracellular lipid signaling molecules as well as components of biomembrane structures in cells, and the biological and/or cellular roles of CerK and C1P were established before and after the identification of human CerK.2–6) in vitro, CerK activity is dependent on divalent ions, particularly calcium ions.1,2,7,8) Treatment with the calcium ionophore A23187 was reported to increase CerK activity and/or C1P formation in A549 carcinoma cells,9) RBL-2H3 cells,10) and CerK-overexpressing Chinese hamster ovary (CHO) cells.11) Thus, Ca2+ ions appeared to be essential for CerK activity or an activator of the enzyme. Cultivation of cells with stimuli such as interleukin-1β9) and serum8) activated CerK activity and/or formation of C1P. Bornancin’s group reported that a cluster of Cys residues (C347XXXC351XXC354) was important for CerK enzyme activity12) and that CerK is a phosphoprotein at least Ser340 and Ser408 residues and phosphorylation of the Ser340 residue impacted stability of the active enzyme conformation.13) Previously, we reported that treatment with orthovanadate, a general inhibitor of tyrosine phosphatase, increased C1P formation via CerK in A549 cells and CHO cells.14) However, precise mechanisms for the post-translational regulation of CerK activity have not yet been elucidated.
The N-terminus of CerK bears a pleckstrin homology (PH) domain, and this domain is indispensable for its activity and acts as a regulator of its targeting and sub-cellular localization.4,15–18) Like many PH domain-containing proteins, CerK can bind with high specificity and affinity to certain phosphatidylinositol (PI) phosphates including PI 4,5-bisphosphate (PI(4,5)P2). However, the effects of glycerophospholipids such as PI, phosphatidylcholine, and PI(4,5)P2 on CerK activity in vitro have not been well established. Cardiolipin (diphosphatidylglycerol), is a unique phospholipid with four acyl chains and two negative charges, and this lipid is generally used to measure CerK activity in vitro.1,19–23) However, it currently remains unknown whether cardiolipin directly interacts with and activates CerK. Thus, we investigated the effects of glycerophospholipids including cardiolipin on recombinant human CerK activity in the glycerol/albumin buffer. In the present study, we showed that cardiolipin bound to CerK and regulated its activity in vitro, and that the modification of cellular cardiolipin levels changed the formation of C1P in intact cells.
The materials used in this study and their sources were as follows. A fluorescent ceramide, 4-nitrobenzo-2-oxa-1,3-diazole-labeled C6-ceramide (NBD-ceramide) was purchased from Molecular Probes (Eugene, OR, U.S.A.); cardiolipin (from bovine heart), glycerol, Triton X-100, and bovine serum albumin (fatty acid-free) were from Sigma-Aldrich (St. Louis, MO, U.S.A.); phosphatidic acid, phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, and PI were from Matreya LLC (Pleasant Gap, PA, U.S.A.); PI(4,5)P2 (1,2-dipalmitoyl) was from Cayman (Ann Arbor, MI, U.S.A.). Stock solutions of the respective lipids were as follows: cardiolipin (ethanol), glycerophospholipids such as phosphatidylcholine and phosphatidylethanolamine (chloroform–methanol, 2 : 1, v/v), PI(4,5)P2 (distilled water), and NBD-ceramide (dimethyl sulfoxide (DMSO)). In some experiments, cells were cultured with cardiolipin micelles in medium with serum. The cardiolipin micelles were prepared by sonication of medium with cardiolipin for 5 min in a bath-type sonicator until the mixture becomes a homogenous dispersion. After being cultured for the indicated times, cells were washed with cardiolipin-free medium and then used to measure the formation of NBD-C1P and cellular levels of cardiolipin.
Construction of Plasmids for Recombinant Human CerK and Green Fluorescent Protein (GFP)-Tagged Phospholipase D6 (PLD6, Mitochondrial PLD) and Preparation of CerKcDNA encoding human CerK (a gift from Dr. Kohama, Daiichi-Sankyo, Co., Ltd., Tokyo, Japan) was amplified by PCR using a forward primer 5′-TAA AGG ATC CAT GGG GGC GAC GGG G-3′, and a reverse primer 5′-TAT AGC TCG AGT CAG CTG TGT GAG TCT GG-3′. This amplification product (full-length, 1–537) was cloned into BamHI and XhoI of pGEX-6P-1 vector to generate the glutathione S-transferase (GST) fusion construct. Likewise, two deletion mutants: PH domain of CerK (1–124, PH-CerK), and CerK lacking PH domain (125–537, ΔPH-CerK) were created using a forward primer 5′-TAA AGG ATC CAT GGG GGC GAC GGG G-3′, and a reverse primer 5′-TAT AGC TCG AGT CAC TCC AGC ATC TCC CG-3′ for PH-CerK, and a forward primer 5′-TAA AGG ATC CAA GCT GAC GTC CAG A-3′, and a reverse primer 5′-TAT AGC TCG AGT CAG CTG TGT GAG TCT GG-3′ for ΔPH-CerK. cDNA encoding human PLD6 (available from Open Biosystems, MHS6278-202808613) was amplified by PCR using a forward primer, 5′-TAT CTC GAG GGC GGC ATG GGA CGG TTG AGT-3′, and a reverse primer 5′-TTA CTG CAG GGT TTG GCT TTC GCT GGA-3′. This amplification product was cloned into the C-terminally GFP-tagged pEGFP-N1 vector at XhoI/PstI sites to generate the GFP fusion construct. GST-fusion proteins were expressed in the E. coli BL21 (DE3) strain upon incubation with 0.05 mM isopropyl 1-thio-β-D-galactoside (IPTG) and 4% ethanol at 25°C for 16 h for full-length CerK and ΔPH-CerK. For PH-CerK expression, the bacteria were incubated with 0.1 mM IPTG and 4% ethanol at 30°C for 4 h. The bacteria were centrifuged at 6000 rpm for 15 min. After the bacterial pellet was re-suspended in lysis buffer (PBS containing 1 mM dithiothreitol, 1% Triton X-100, 10% glycerol, 0.5 mM phenylmethylsulfonyl fluoride (PMSF)), the lysates were sonicated. GST-fusion proteins were collected on glutathione-Sepharose beads from the lysates, washed five times with lysis buffer, and eluted with elution buffer (20 mM reduced glutathione, 50 mM Tris–HCl, pH 8.0).
CerK Activity AssayThe activity of recombinant CerK in vitro with 10 µM NBD-ceramide was measured in the two buffers, as described previously20,22) with modifications. The glycerol/albumin buffer was used to examine the effects of glycerophospholipids including cardiolipin on CerK activity. As described in the Results, supplementation of glycerophospholipids including cardiolipin did not change CerK activity in the cardiolipin/Triton X-100 buffer. To study the CaCl2/MgCl2 responsibility on CerK activity, both the glycerol/albumin buffer and cardiolipin/Triton X-100 buffer were used. The assays using the glycerol/albumin buffer were performed as follows. NBD-ceramide and cardiolipin were directly added to the reaction buffer (90 µL). CerK (30 ng/tube, 10 µL) was added to the assay mixtures (total volume 100 µL), and incubated at 37°C for 15 min. The assay mixture consisted of 10% glycerol, 0.02% albumin, 100 mM KCl, 1 mM dithiothreitol, 1 mM ATP, 40 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES) (pH 7.4), CaCl2, and/or MgCl2 at the indicated concentrations. The assays using the cardiolipin/Triton X-100 buffer were performed as follows. Mixed micelles containing cardiolipin, Triton X-100, and NBD-ceramide were prepared by drying lipids from stock solutions with N2 gas and then sonicating (3 min) in reaction buffer (100 mM KCl, 1 mM dithiothreitol, 1 mM ATP, 40 mM HEPES, pH 7.4). The micelles were diluted five-fold with reaction buffer, and 100-µL reactions were initiated by the addition of CerK (30 ng), CaCl2, and/or MgCl2. The final concentrations of the reagents in the mixed micelles were: 36 µM cardiolipin, 0.0125% Triton X-100, and 10 µM NBD-ceramide. In the two assays, concentration of glutathione derived from CerK preparation was 0.046 mM. In both buffers, the reaction was stopped by the addition of 125 µL of chloroform and 250 µL of methanol after the incubation. In order to extract C1P to the organic phase, the pH of the mixture was adjusted to approximately 2–3 by the addition of 5 N HCl (1 µL), and the mixtures were vigorously vortexed and incubated at 4°C for 10 min. After adding 125 µL of chloroform and 125 µL of water, the samples were then divided into organic and aqueous phases using centrifugation (3000 rpm, 10 min). NBD-C1P extracted in the organic phase (100 µL) was dried under nitrogen. Dried samples were dissolved in 10 µL of chloroform–methanol (1 : 1) and analyzed on silica gel-60 TLC plate (Merk, Germany). The formation of NBD-C1P was measured for 15 min at 37°C, and CerK activity (pmol NBD-C1P/µg CerK protein/min) was examined. In the glycerol/albumin and cardiolipin/Triton X-100 buffers with and without supplements, such as CaCl2–MgCl2 and the indicated lipids, the formation of NBD-C1P was linear for 30 min depending on the concentration of CerK from 10 to 30 ng per assay tube.
Lipid–Protein Binding Assays: Overlay Assay and Large Multilamellar Vesicle Binding AssayAssays were performed as previously described24) with minor modifications. Briefly, tested lipids were dissolved in the indicated solvents and spotted onto a Hybond C membrane (Amersham) in the lipid–protein overlay assay. After drying, the membrane was rewetted and blocked for 1 h in the TBS/Tween 20 buffer containing 2% albumin, and then exposed overnight at 4°C to respective preparations of GST-tagged recombinant human CerK proteins containing 0.1 mM CaCl2. In some cases, the CerK preparations containing 1 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) were used. The washed membranes were treated with an anti-GST antibody (27457701 V, GE Health Care, Buckinghamshire, U.K.), and the immunoreactive spots were visualized using a second antibody. In the vesicle binding assay, vesicles of cardiolipin or phosphatidylcholine were prepared by vigorously vortexing the lipids in the buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.1 mM CaCl2). The binding reaction was initiated by the adding of lipid vesicle solutions and GST-CerK. After 5 min at room temperature, the reaction mixture was centrifuged at 17000×g for 10 min, and the supernatant was removed. After repeated washing and centrifugation, the pellet was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and the immunoreactive bands to an anti-GST antibody were visualized using a second antibody. In the two assays, purified recombinant GST did not bind to any of the lipids.
Transfection of HepG2 Cells with the PLD6-GFP Vector, and Measurement of PLD6-GFP ExpressionRegarding protein expression, cells were seeded at a density of 4.5×104 cells/well (12-well plate), cultured for 2 d in Dulbecco’s modified Eagle’s medium (DMEM) with serum, and transiently transfected with 2 µg of expression vectors for Lipofectamine PLUS (Invitrogen) in Opti-MEM without serum, according to the manufacturer’s protocol. After a 3-h incubation, transfected cells were further cultured with DMEM with serum for 24 h, and then used in various assays such as the expression of PLD6-GFP, measurement of intracellular cardiolipin levels, and formation of C1P. The expression of PLD6-GFP was confirmed by Western blotting using an anti-GFP antibody (sc-8334, Santa Cruz Biotech. Dallas, TX, U.S.A.). In some cases, transfected cells were seeded on the coverslips (12 mm in diameter) of glass-bottomed dishes (Iwaki, Tokyo, Japan). After being cultured for 40–48 h, the cells were washed with buffer and GFP-derived fluorescent images were taken with a FluoView-FV500 confocal laser scanning microscope system (Olympus, Tokyo).
Measurement of Intracellular Cardiolipin LevelsPhospholipids including cardiolipin in HepG2 cells expressing PLD6-GFP and cultured with cardiolipin were extracted in chloroform–methanol–HCl solution as described previously25) with minor modifications. The organic fraction after evaporation was separated on silica gel-60 HPTLC plate. The positions of phospholipids such as cardiolipin and PI on the plate after visualization using iodine staining were determined by matching the migration distance to standards and previously reported Rf values. The intensity of iodine staining was measured using NIH ImageJ software.
Measurement of NBD-C1P Formation in HepG2 CellsCells were incubated with 10 µM NBD-ceramide for 30 min or 1 h, and lipids including NBD-labeled ceramide metabolites in wells (both cells and medium) were extracted by chloroform–methanol. NBD-labeled ceramide metabolites including NBD-C1P were separated using TLC, and the levels of metabolites were determined as described previously.14,26,27) Absolute values (pmol/well) of ceramide metabolites in HepG2 cells were similar to those in cells previously reported,14,26,27) and the relative levels of NBD-sphingomyelin, NBD-glucosylceramide, and NBD-C1P at 30 min after labeling with NBD-ceramide were 5–10 (% of NBD-ceramide), 3–6, and 0.05–0.1%, respectively. Since the ceramide metabolites showed a wide range of values and the absolute values varied depending on experiments, the data in Figs. 4E and 5B were expressed as percentages of the respective metabolites in control cells.
Analysis of Cell Morphology and Cell DetachmentCellular effects induced by modification of cardiolipin levels were examined by cell morphology and cell detachment. The morphological changes of cells at 50–60% confluence were observed by differential interference contrast microscopy. Cells were cultured with serum at 70–80% confluence, and cell numbers attached on plates were counted at 24 h after cardiolipin treatment.
Data Presentation and StatisticsValues are the mean±standard deviation (S.D.) for the indicated number (n) of independent experiments. The values in Fig. 5 are the mean±standard error of the mean (S.E.M.) of three independent experiments performed in duplicate. In the case of multiple comparisons, the significance of differences was determined using a one-way analysis of variance with Dunnett’s or Tukey’s test. The Student’s two-tailed t-test was used for pairwise comparisons. p Values <0.05 were considered to be significant.
First, activities of human recombinant CerK were examined with and without CaCl2 and MgCl2. In the glycerol/albumin buffer containing the substrates (1 mM ATP and 10 µM NBD-ceramide), our preparations of recombinant CerK exhibited a marked activity in vitro (Figs. 1A, B, open circles). The activity was 44.1±11.4 pmol/µg/min (n=7, Fig. 1C). CerK activity was increased by the addition of CaCl2 and MgCl2, and the addition of CaCl2 and MgCl2 at 0.1 mM led to almost maximal stimulatory effects on CerK activity (Fig. 1C). CaCl2 and MgCl2 at 2 mM both increased this activity approximately 3-fold over that of control activity. CerK activity was previously reported to increase from 0.1 to 10 µM Ca2+,1,3) thus, we investigated the roles of CaCl2 on CerK activity in detail. The activities of CerK with and without 0.1 mM CaCl2 were almost completely abolished in the presence of 2 mM EGTA, while those with 2.5 mM CaCl2 and 0.1 mM MgCl2 were not modified by EGTA (Fig. 1D). The addition of not only 2 mM EGTA, but also 2 mM ethylenediaminetetraacetic acid (EDTA) to the assay mixtures markedly decreased CerK activity (Table 1). The addition of CaCl2 at 10 µM did not increase CerK activity (40–50 pmol/µg/min), whereas 50 µM CaCl2 markedly increased CerK activity to approximately half of that with 0.1 mM. The combination of CaCl2 and MgCl2 at 1 mM did not enhance CerK activity; the value with CaCl2/MgCl2 were 0.97±0.12-fold of that with MgCl2 alone (n=3).
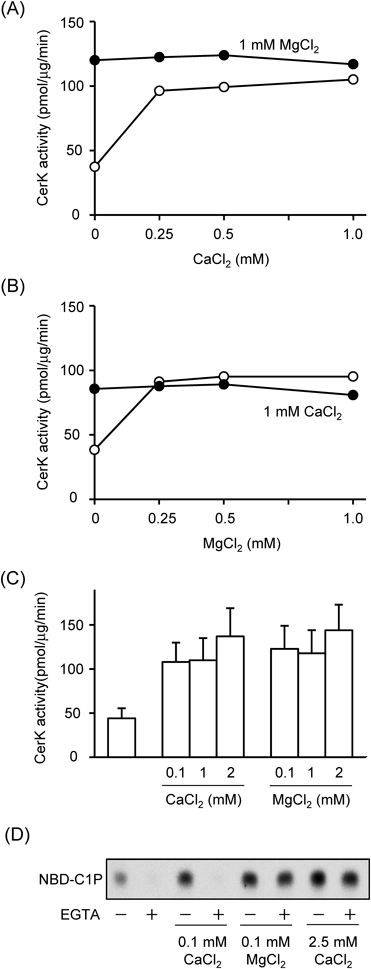
CerK activity was measured with the indicated concentrations of CaCl2 or MgCl2. In (A) and (B), CerK activity was examined with CaCl2/MgCl2 alone (open circles) and in the presence of MgCl2 or CaCl2 at 1 mM (closed circles). Data are from a typical experiment, and images of TLC analyses were shown in Supplementary Fig. 1. Quantitative data are shown in (C). In (D), CerK activity was measured with the indicated concentrations of CaCl2/MgCl2. In some cases, assay mixtures were further supplemented with 2 mM EGTA. Data were from a typical representative experiment. Quantitative data for EGTA on CerK activity are shown in Table 1.
| Buffers | None | EGTA | EDTA |
|---|---|---|---|
| CerK activity (%) | |||
| Glycerol/Albumin | 100 | 16.7±8.6 | 2.1±0.5 |
| Cardiolipon/Triton X-100 | 100 | 21.2±9.6 | 2.4±0.4 |
CerK activity was measured with 2 mM EGTA or 2 mM EDTA in the glycerol/albumin buffer and cardiolipin/Triton X-100 buffer. CaCl2 and MgCl2 were not added to the reaction buffer in this experiment. Data were expressed as a percentage of control activity without EGTA/EDTA in the respective buffer, and are the mean±S.D. of three independent experiments.
Next, we investigated the effects of various glycerophospholipids on CerK activity in the glycerol/albumin buffer. Phosphatidylcholine, phosphatidylethanolamine, PI, and phosphatidylserine at 0.1 µM had no significant effect on CerK activity (Table 2). Phosphatidic acid, an acidic glycerophospholipid, at 0.1 µM did not change CerK activity. These lipids including phosphatidic acid at 1 µM did not change CerK activity; the values of CerK activity with the lipids were from 0.9- to 1.1-fold and from 3.1- to 3.4-fold of control in the absence and presence of 0.1 mM CaCl2, respectively, which were similar as those without the lipids. PI(4,5)P2 at 0.1 µM had no effect, whereas the lipid at 1 µM slightly inhibited CerK activity with 0.1 mM CaCl2 (Table 2): a relative CerK activity with 1 µM PI(4,5)P2 was 82±8% (% of the control without the lipid, n=3). Addition of 0.1 µM cardiolipin (diphosphatidylglycerol) slightly stimulated CerK activity in the absence of CaCl2, but the responses were not significant (Figs. 2A, B). In the presence of 0.1 mM CaCl2, cardiolipin showed a bell-shaped response that depended on the lipid concentrations: cardiolipin stimulated CerK activity from 0.05 to 0.1 µM and inhibited it at concentrations greater than 0.5 µM. The activities of CerK with 1 and 5 µM cardiolipin were significantly less than that with 0.1 µM. Cardiolipin at 0.1 µM increased CerK activity with 0.1 mM MgCl2 (Figs. 2C, D). In the presence of 0.1 mM MgCl2, the activity of CerK with 1 µM cardiolipin was 1.6- and 1.8-fold in two independent experiments, which were markedly less than that with 0.1 µM. Thus, cardiolipin appeared to show dual effects, stimulation with 0.1 µM and inhibition with 1 µM, on CerK activity in the presence of MgCl2. We examined the effect of 0.1 µM cardiolipin on the concentration-dependency of CaCl2 or MgCl2 from 0.01 to 0.1 mM, and the lipid did not affect the sensitivity of CerK activity to CaCl2 and MgCl2 in a typical experiment (data not shown). These results demonstrated that cardiolipin, not other glycerophospholipids tested, directly affected CerK activity in vitro in the glycerol/albumin buffer containing CaCl2 or MgCl2.
| None | CaCl2 | |
|---|---|---|
| CerK activity (fold) | ||
| Vehicle | 1 | 3.2±0.4 |
| PA | 1.2±0.4 | 3.8±0.3 |
| PC | 1.2±0.3 | 3.3±0.3 |
| PE | 1.1±0.2 | 3.4±0.4 |
| PI | 1.1±0.2 | 3.2±0.3 |
| PS | 1.2±0.2 | 3.5±0.3 |
| PI(4,5)P2 | 1.2±0.2 | 3.4±0.4 |
| PI(4,5)P2 (1 µM) | 1.1±0.1 | 2.7±0.2 |
CerK activity was measured in the glycerol/albumin buffer with and without 0.1 mM CaCl2. Vehicle, phosphatidic acid (PA), phosphatidylcholine (PC), phosphatidylethanolamine (PE), PI, phosphatidylserine (PS), and PI(4,5)P2 at 0.1 µM were added to the assay mixtures. In some cases, the effect of 1 µM PI(4,5)P2 was examined. CerK activity was expressed as a fold of the control without CaCl2. Data are the mean±S.D. of three or four independent experiments.

In (A)–(D), CerK activity was measured with the indicated concentrations of cardiolipin in the glycerol/albumin buffer. In some cases, assay mixtures were further supplemented with 0.1 mM CaCl2 (A, B) or 0.1 mM MgCl2 (C, D). Typical images of TLC analyses were shown in (A) and (C), and quantitative data were shown in (B) and (D). Data were expressed as a fold of the control without CaCl2/MgCl2, and are the mean±S.D. of three independent experiments. * p<0.05, significantly different between the two groups. In (E), CerK activity was measured with the indicated concentrations of CaCl2 or MgCl2 in the cardiolipin/Triton X-100 buffer. Data are the mean±S.D. of three independent experiments.
Next, we examined CerK activity in the cardiolipin/Triton X-100 buffer, and the basal activity and the CaCl2–MgCl2 responsibility in the buffer were compared with those in the glycerol/albumin buffer (Fig. 2E, Table 3). CerK activity was 26.7±2.9 pmol/µg/min (n=7) without the exogenous addition of CaCl2/MgCl2 which was less than that in the glycerol/albumin buffer, 44.1±11.4 pmol/µg/min. When CerK activity of a same recombinant CerK preparation was measured in the two buffers in an experiment, CerK activity in the cardiolipin/Triton X-100 buffer was 40.1±9.2% of that in the glycerol/albumin buffer (p<0.05, n=3). CerK activity in the cardiolipin/Triton X-100 buffer was increased by the addition of CaCl2 and MgCl2. Interestingly, the CerK activity with 1 mM MgCl2 in the cardiolipin/Triton X-100 buffer was two-fold of that in the glycerol/albumin buffer. The addition of CaCl2 at 10 µM did not increase CerK activity (20–30 pmol/µg/min), and the activity with 50 µM CaCl2 was approximately half of that with 0.1 mM in the cardiolipin/Triton X-100 buffer. The CerK activity with CaCl2/MgCl2 at both 1 mM was 0.84±0.21-fold of that with MgCl2 alone (n=3). Similar to the response in the glycerol/albumin buffer, CerK activity was markedly decreased in the presence of 2 mM EGTA and EDTA in the cardiolipin/Triton X-100 buffer (Table 1). In the cardiolipin/Triton X-100 buffer, supplementation of glycerophospholipids such as PI(4,5)P2 and cardiolipin at concentrations from 0.1 to 1 µM did not change CerK activity (data not shown). The obtained data, regulation of CerK activity by cardiolipin in the in the glycerol/albumin buffer and a greater CerK activity in the cardiolipin/Triton X-100 buffer with MgCl2 showed that cardiolipin affected the activity of human recombinant CerK.
| Buffers | Glycerol/Albumin | Cardiolipin/Triton X-100 |
|---|---|---|
| CerK activity (pmol/µg/min) | ||
| None | 44.1±11.4 | 26.7±2.9 |
| 1 mM CaCl2 | 110±25 | 101±12 |
| 1 mM MgCl2 | 118±26 | 240±11 |
| CaCl2 and MgCl2 | CerK activity (fold of the value with MgCl2) | |
| 0.97±0.12 | 0.84±0.21 | |
Some data shown in Fig. 1C (the glycerol/albumin buffer) and Fig. 2E (cardiolipin/Triton X-100 buffer) were summarized. CerK activity was measured with vehicle, 1 mM CaCl2, 1 mM MgCl2, and the combination of CaCl2/MgCl2. The effects of combination of CaCl2/MgCl2 were expressed as a percentage of activity with 1 mM MgCl2 in the respective buffer, and are the mean±S.D. of three independent experiments.
We employed another method to show the direct effect of cardiolipin on CerK: lipid–protein binding assays in two manners. In the lipid–protein overlay assay, cardiolipin bound to GST-CerK in a lipid concentration-dependent manner in the presence of 0.1 mM CaCl2 (Figs. 3A, B). PI(4,5)P2 and phosphatidic acid at 2 nmol also bound to GST-CerK, as described previously.16,17) Other glycerolipids tested did not bind to GST-CerK. Cardiolipin bound to GST-ΔPH-CerK (Fig. 3C), but not GST-PH-CerK (Fig. 3D) or the GST protein (Fig. 3E), under our conditions. Since divalent cations including Ca2+ may interact with the negatively charged head-group of cardiolipin, we examined the effects of CaCl2 on cardiolipin binding to CerK. The direct binding of cardiolipin to GST-CerK and GST-ΔPH-CerK was observed in the presence of 1 mM EGTA without CaCl2: the bindings of both proteins were approximately 80–90% of those with 0.1 mM CaCl2. The direct binding of cardiolipin to GST-CerK was examined in another lipid–protein binding assay, the large multilamellar vesicle binding assay (Fig. 3F). CerK bound to vesicles containing cardiolipin, but not phosphatidylcholine.
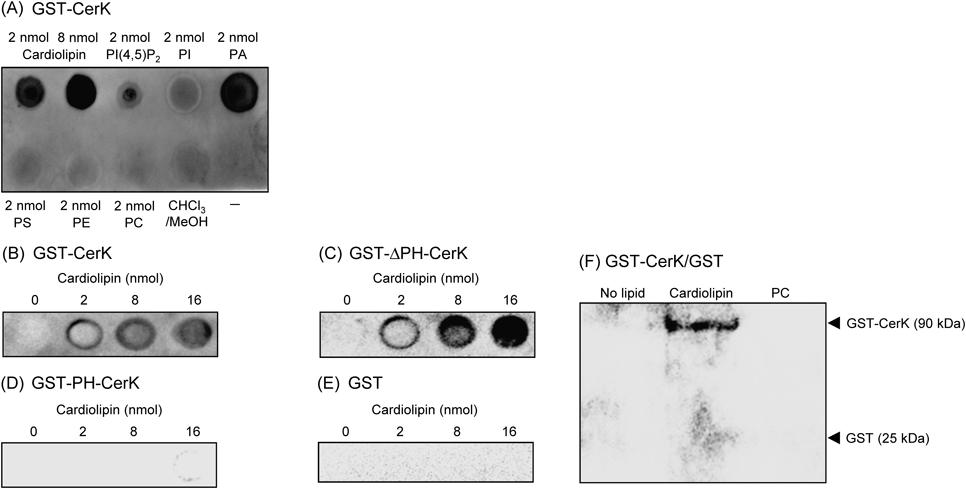
The indicated amounts of cardiolipin, lipids, and the solvent were spotted onto a Hybond C membrane, and the lipid–protein overlay assay was performed. GST-CerK (A, B), GST-ΔPH-CerK (C), GST-PH-CerK (D), and GST (E) were used. In (F), the large multilamellar vesicle binding assay was performed. Vesicles of cardiolipin and phosphatidylcholine (PC) were incubated with GST-CerK and GST. Data are representative of three independent experiments.
Choi et al. reported that over-expression of mitochondrial PLD6 caused a marked decrease in cardiolipin levels and that PLD6-GFP localized in mitochondria in NIH3T3 cells.25) Therefore, we attempted to regulate cardiolipin levels in HepG2 cells by transient transfection with the vector for PLD6-GFP (Fig. 4). The expression of PLD6-GFP was confirmed at the protein level (Fig. 4A). Choi et al. showed the localization of PLD6-GFP in the mitochondria in NIH3T3 cells.25) A similar intracellular localization of PLD6-GFP was observed in HepG2 cells (Fig. 4B). The cellular amounts of cardiolipin, but not those of other glycerophospholipids such as phosphatidylcholine and PI, were decreased in cells expressing PLD6-GFP (Fig. 4C). In cells expressing PLD6-GFP, cardiolpin level was 71% (% of the control), and levels of PE, PI, PS, and PC were 98, 99, 102, and 103%, respectively, in a typical experiment. Control HepG2 cells and HepG2 cells expressing PLD6-GFP were incubated with 10 µM NBD-ceramide for 30 min, and the levels of NBD-fluorescence in the extracted cellular lipids were analyzed. The formation of NBD-C1P was significantly less in HepG2 cells expressing PLD6-GFP than in the control cells (Figs. 4D, E). The formation of NBD-sphingomyelin was slightly but significantly less in HepG2 cells expressing PLD6-GFP than in control cells. Cellular levels of NBD-glucosylceramide, NBD-ceramide, and the endogenous CerK protein (data not shown) were almost the same in the two cells. These results suggest a probable stimulatory effect of endogenous cardiolipin on CIP formation. In contrast, cultivation of HepG2 cells with cardiolipin at greater concentrations over 10 µM for 24 h showed an inhibitory effect on the formation of NBD-C1P (Fig. 5). The cardiolipin treatment did not appear to change amounts of NBD-ceramide taken up and other ceramide metabolites: for instance, the values of NBD-ceramide, NBD-glucosylceramide and NBD-sphingomyelin in 50 µM caldiolipin-treated cells were 96–104% of the respective values in control cells. In a typical experiment, the levels of cardiolipin including the lipid attached to the cell membrane in HepG2 cells treated for 24 h with 50 µM cardiolipin were 120–130% those of control cells without cardiolipin. The levels of other phospholipids such as phosphatidylcholine and PI were not changed by the cultivation of cells with cardiolipin (data not shown). We could not detect the stimulatory effects of cardiolipin exogenously added under our experimental conditions: the levels of NBD-C1P in HepG2 cells incubated for 1 h with NBD-ceramide in the presence of 0.1–10 µM cardiolipin were approximately 90% that of control cells without cardiolipin. The cultivation of HepG2 cells expressing PLD6-GFP for 24 h with 0.1–10 µM cardiolipin did not reverse the decreased formation of C1P, and we could not examine the effects of caldiolipin at greater concentrations over 10 µM on the cells because the cultivation caused cell detachment (data not shown). Over-expression of PLD6-GFP by itself and application of cardiolipin alone from 10 to 50 µM for 24 h did not cause cell rounding and cell detachment (data not shown).
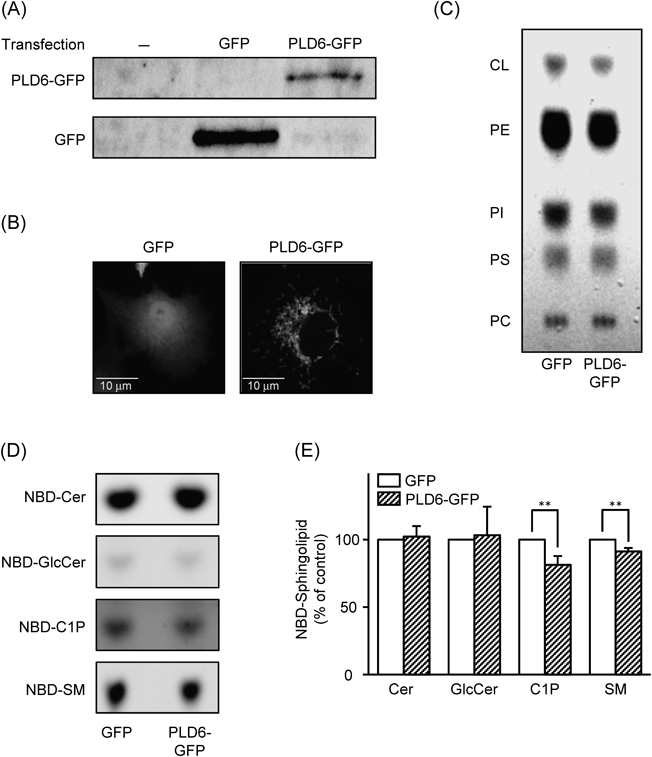
HepG2 cells were transiently tranfected with expression vectors for GFP and GFP-tagged PLD6, and cultured for 24 h. (A) The expression of GFP and PLD6-GFP proteins was confirmed by Western blotting. (B) Green-fluorescence derived from the PLD6-GFP protein in cells was confirmed by confocal laser microscopy. (C) The phospholipids in cells were extracted, and separated on a TLC plate. Cardiolipin (CL), phosphatidylethanolamine (PE), PI, phosphatidylserine (PS), and phosphatidylcholine (PC). Data in (A), (B), and (C) are representative of two independent experiments. In (D) and (E), cells expressing GFP and PLD6-GFP were incubated with 10 µM NBD-ceramide for 30 min, and ceramide metabolites including NBD-C1P in wells (cells and medium) were extracted. Ceramide metabolites were separated by a TLC method. Typical images of NBD-ceramide and the metabolites on TLC plates were shown in (D), and quantitative data were shown in (E). NBD-ceramide (NBD-Cer, Cer), NBD-glucosylceramide (NBD-GlcCer, GlcCer), NBD-C1P (C1P), NBD-sphingomyelin (NBD-SM, SM). Data were expressed as percentages of respective NBD-labeled molecules in control cells expressing GFP alone. Data are the mean±S.D. of three independent experiments. ** p<0.01, significantly different from the control.
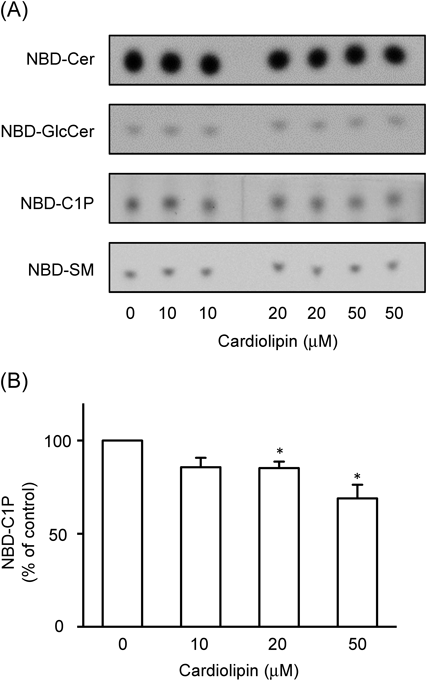
HepG2 cells were cultured with cardiolipin at the indicated concentrations for 24 h, and the cells after washing were incubated with 10 µM NBD-ceramide for 1 h. Typical images of NBD-ceramide metabolites on the TLC plate were shown in (A), and quantitative data for NBD-C1P formation were shown in (B). Data were expressed as a percentage of NBD-C1P levels in the control cells, and are the mean±S.E.M. of three independent experiments performed in duplicate. * p<0.05, significantly different from the control.
In the present study, we demonstrated that cardiolipin bound to CerK in vitro and showed diverse effects on CerK activity in vitro and in intact cells (a probable stimulation by endogenous cardiolipin and inhibition by exogenously added cardiolipin).
Binding of Cardiolipin to CerK in VitroCardiolipin is used as a molecule-stabilizing reagent in various assay systems in vitro, and has been shown to interact with various proteins possibly through the physical properties of the anionic lipid.28–30) Also, activity of CerK was measured in cardiolipin-containing buffers in many previous reports.1,19,20,22,23) However, direct evidence to show the physical association between cardiolipin and CerK as well as activation of this enzyme by the lipid does not currently exist. In the present study, we showed for the first time that cardiolipin directly bound to recombinant CerK in vitro using two lipid–protein binding assays, an overlay assay and large multilamellar vesicle binding assay (Fig. 3). In the lipid–protein overlay assay, cardiolipin bound to the full length of CerK and ΔPH-CerK, but not PH-CerK. Previous studies reported that ΔPH-CerK has the capacity to bind to lipids. Carré et al. reported that ΔPH-CerK exhibited slight, but distinct binding to liposomes of phospholipids containing phosphatidylethanolamine, phosphatidylcholine, and phosphatidylserine in a sucrose-loaded liposome assay; however, PH-CerK strongly bound to the liposomes.15) Kim et al. reported that PI(4,5)P2 mainly bound to PH-CerK, but other poly-phosphorylated PIs including PI 3-phosphate bound to CerK via the non-PH domain of CerK.16) Rovina et al. reported that PI(4,5)P2 bound to the C-terminally FLAG-tagged CerK, but not to the C-terminally FLAG-tagged PH domain of CerK, in a similar lipid–protein overlay assay.17) In addition to PH domain, poly-phosphorylated forms of PI including PI(4,5)P2 have been shown to bind to various domains having basic motifs in proteins.31) These results including ours suggest that CerK may have multiple binding domains of the lipids such as cardiolipin and PI(4,5)P2. The exact domains and/or amino acids of CerK responsible for cardiolipin binding have yet to be identified.
Regulation of CerK Activity by Cardiolipin in VitroBoth PI(4,5)P2 and phosphatidic acid were shown to bind to CerK.16,17) It was reported that PI(4,5)P2 did not change CerK activity,16) while the effect of phosphatidic acid on the activity has not yet been demonstrated. We confirmed the binding of CerK to PI(4,5)P2 and phosphatidic acid (Fig. 3A). Under our conditions, supplementation with PI(4,5)P2 and phosphatidic acid at 0.1 µM did not change CerK activity, although 1 µM PI(4,5)P2 slightly inhibit the activity. Other glycerophospholipids including PI and phosphatidylcholine did not change CerK activity with or without 0.1 mM CaCl2 (Table 2). Interestingly, we found that cardiolipin had diverse effects on CerK activity in vitro: cardiolipin at 0.1 µM significantly increased CerK activity, whereas the lipid at concentrations greater than 1 µM had inhibitory effects in the glycerol/albumin buffer. Thus, cardiolipin appeared to be the first case for the direct regulation of CerK by glycerophospholipids. The concentration dependency of CaCl2 and MgCl2 in the cardiolipin/Triton X-100 buffer (Fig. 2E) was almost the same as that in the glycerol/albumin buffer (Fig. 1C), and the addition of cardiolipin did not appear to change this dependency in the glycerol/albumin buffer. CerK activity without CaCl2/MgCl2 in the cardiolipin/Triton X-100 buffer was significantly less than that in the glycerol/albumin buffer, and its activity with 1 mM CaCl2 was almost the same in the two buffers (Figs. 1C, 2E). Thus, cardiolipin at concentrations that had a stimulatory effect on CerK appeared to increase intrinsic activity of the enzyme. CerK activity showed a greatest value with MgCl2 in the cardiolipin/Triton X-100 buffer containing 36 µM cardiolipin, and the lipid at 0.1 µM increased CerK activity with MgCl2 in the glycerol/albumin buffer. Although it was difficult to compare the concentration dependency of cardiolipin in the two buffers because of Triton X-100 and/or glycerol, caldiolipin appeared to increase CerK activity in vitro. Cardiolipin was reported to increase intrinsic activity of mitochondrial-associated neutral sphingomyelinase.32) It remains to be solved how cardiolipin acts as a bi-directional regulator of CerK activity in vitro (and in intact cells). In the present study, we used cardiolipin extracted from bovine heart. Alteration in its acyl chain composition and peroxidation of cardiolipin have been proposed to show different effects in tissues-dependent manner.33,34) Precise mechanisms to explain the biphasic regulation of CerK and/or C1P formation by cardiolipin including specificity of molecular forms of cardiolipin remain to be elucidated.
Regulation of CerK Activity by Cardiolipin in CellsIn the present study, a deviation from the normal range of cardiolipin, both the decrease by PLD6 expression and increase by an exogenous addition of the lipid, negatively regulated C1P formation in intact cells, although we could not have a direct evidence showing a stimulatory effect of cardiolipin. CerK was mainly detected in the plasma membranes, Golgi complex, and endosomes in cells,1,4,19,21) although the localization of CerK in the cytosol was reported in rat basophilic leukemia (RBL-CK3) cells.10) CerK appeared to be attached to the cytosolic face of the cellular organelles including the plasma membranes.4,21) Cardiolipin, which is mainly formed in the matrix of mitochondria and is, thus, extensively found in mitochondrial membranes. How and/where cardiolipin interacts with CerK in cells ? In the present study, we showed that the formation of NBD-C1P was reduced in HepG2 cells expressing mitochondrial PLD6 and showing low cardiolipin levels (Fig. 4). Since the CerK protein and its activity have been shown to exist, at least partially, within mitochondria in A549 lung adenocarcinoma cells,21) cardiolipin existing endogenously in cells may stimulate CerK in the mitochondria. However, we could not observe a marked mitochondrial association of the C-terminally GFP-tagged CerK in HepG2 cells, like in CHO cells.35) The pre-treatment of HepG2 cells with cardiolipin for 24 h significantly inhibited the formation of NBD-C1P (Fig. 5). Exogenously added cardiolipin was shown to move to mitochondria through a fusion process between this lipid and the plasma membrane in glioblastoma cells,36) and the dynamic cellular movement of cardiolipin has been proposed between mitochondria and other organelles including endosomes in cells.28,37) Enzymes for ceramide-related pathways including sphingosine kinases 1/2 were activated by cardiolipin in vitro,32,38–43) and many enzymes of the pathway exist in non-mitochondrial compartments such as the plasma membrane and endosomes. A probable intracellular movement of cardiolipin and/or a transient localization of cardiolipin may explain the interaction of ceramide metabolic enzymes including CerK and cardiolipin in the non-mitochondrial compartments in cells. Identification of intracellular compartments for cardiolipin–CerK interaction remains to be elucidated. Alterations of cardiolipin in cells have been shown to be involved in various pathological conditions including ischemia, hypothyroism, Barth syndrome, and aging, etc.33,34) Also, CerK and C1P appear to be involved in ischemia and neurodegenerative diseases.41,42) Possible interactions between cardiolipin and CerK/C1P should be determined physiologically and pathologically.
Effects of Ca2+ and Mg2+ on CerK ActivityIn the present study, the addition of CaCl2 increased CerK activity in both the glycerol/albumin and cardiolipin/Triton X-100 buffers (Figs. 1, 2). The addition of 2 mM EGTA markedly decreased CerK activity with 0.1 mM CaCl2 without changing its activity with 0.1 mM MgCl2 in the glycerol/albumin buffer (Fig. 1D). These results suggested the selective trapping of Ca2+ by EGTA and were consistent with previous findings showing that CerK was a Ca2+-dependent enzyme.1–3) Under our conditions, the addition of CaCl2 up to 10 µM did not increase CerK activity in either buffer. Similar findings were reported in immunoprecipitated CerK activity from rat basophilic leukemia cells.10) The addition of MgCl2 increased CerK activity without CaCl2 in both buffers (Figs. 1C, 2E). Since the addition of 0.1 mM MgCl2 increased CerK activity in the presence of EGTA, MgCl2 appeared to have stimulatory effects on CerK activity in a Ca2+-independent manner. The results obtained in vitro suggested that either CaCl2 or MgCl2, possibly the divalent cations, Ca2+ or Mg2+, was essential for CerK activity. In the cardiolipin/Triton X-100 buffer, the addition of 1 mM MgCl2 alone increased CerK activity by 8-fold, which was much greater than that by 1 mM CaCl2, and the co-addition of CaCl2–MgCl2 did not show additive and/or synergic effects. The results were consistent with those in a previous report by Van Overloop et al.19) In kinase reactions, divalent cations such as Mg2+ and Ca2+ interact and reduce negative charges in the phosphate groups of ATP, and may generally enhance the transfer of phosphate groups to substrates and the association between enzymes and ATP. Previous findings and the present results suggest that Ca2+ is not a direct activator of CerK, while divalent cations such as Mg2+ and Ca2+ appear to be essential factors for a kinase reaction of CerK. Mitsutake and Igarashi reported that calmodulin bound to CerK, which resulted in the Ca2+-dependent activation of CerK.43) The regulatory mechanisms of Ca2+ on CerK including roles of Ca2+-binding proteins such as calmodulin and calcineurin on the enzyme have yet to be established.
We thank Dr. Takafumi Kohama (Daiichi-Sankyo, Tokyo, Japan) and Prof. A. Nishida (Chiba University, Japan) for providing the vector for human CerK and NBD-ceramide, respectively. These studies were partially supported by Grants-in-Aid (24790066, 26460060 to H.N. and 26460093 to T.M.) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
The authors declare no conflict of interest.
The online version of this article contains supplementary materials.