Abstract
We investigated the effect of chikusetsusaponin IVa (CS) and chikusetsusaponin IVa methyl ester (CS-ME) from the roots of Achyranthes japonica NAKAI on lipopolysaccharide (LPS)-induced nitric oxide (NO) and prostaglandin E2 (PGE2) production in RAW264.7 macrophages. CS-ME more potently inhibited LPS-induced NO and PGE2 production than CS. CS-ME concentration-dependently inhibited LPS-induced tumor necrosis factor (TNF)-α and interleukin (IL)-6 and IL-1β production in RAW264.7 macrophages and mouse peritoneal macrophages. Consistent with these findings, CS-ME suppressed LPS-induced expression of inducible NO synthase (iNOS) and cyclooxygenase (COX)-2 at protein level as well as iNOS, COX-2, TNF-α, IL-6, and IL-1β at mRNA level. In addition, CS-ME suppressed LPS-induced transcriptional activity of nuclear factor (NF)-κB and activator protein (AP)-1. The anti-inflammatory properties of CS-ME might result from suppression of iNOS, COX-2, TNF-α, IL-6, and IL-1β expression through downregulation of NF-κB and AP-1 in macrophages.
Chronic inflammation is an undesirable phenomenon that can ultimately result in inflammatory diseases, such as arthritis, asthma, multiple sclerosis, inflammatory bowel disease, and atherosclerosis.1,2) Macrophages detect pathogenic substances through pattern-recognition receptors (PPR) including toll-like receptors (TLRs), and subsequently regulate inflammatory response3) using a wide range of soluble pro-inflammatory mediators, such as, nitric oxide (NO), prostaglandin E2 (PGE2), tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), and IL-1β. Over-produced pro-inflammatory mediators further aggravate the inflammatory infiltration and immune responses in many acute and chronic inflammatory diseases.4) Lipopolysaccharide (LPS) is one of the most powerful activators of macrophages known, and activation of macrophages by LPS is resulting from TLR4-mediated intracellular signaling cascades, such as nuclear factor (NF)-κB, activator protein-1 (AP-1), signal transducers and activators of transcription (STATs), and interferon response factors (IRFs).4) NF-κB is mainly composed of p50 and p65.5) AP-1 functions in dimeric forms consisting of Fos (c-Fos, FosB, Fra-1, Fra-2) and Jun (c-Jun, v-Jun, JunB, JunD) subfamilies.6) In response to LPS, NF-κB and AP-1 proteins translocate to the nucleus, which binds to the consensus DNA sequence and induce expressions of pro-inflammatory mediators.7)
Achyranthes japonica NAKAI (Amaranthaceae) is a perennial herb and widely distributed throughout in Korea. Its roots have been called “Soemoo-reup” in Korea, which means ox knee derived from their shape.8) It has been reported that the roots of A. japonica exhibit various pharmacological effects, such as anti-inflammatory and antioxidative effects.8,9) In our search for anti-inflammatory active compounds from A. japonica, we isolated two triterpenoid saponins, chikusetsusaponin IVa (CS) and chikusetsusaponin IVa methyl ester (CS-ME) (Fig. 1). CS and CS-ME have been reported to possess anti-tumor,10) anti-diabetic,11) antihyperglycemic,12) and anti-thrombotic activities.13) However, no report has previously been published on their anti-inflammatory effects and related mechanisms. Therefore, as a part of our ongoing screening program to evaluate the anti-inflammatory potentials of natural compounds, we investigated the cell-based anti-inflammatory effects of CS and CS-ME in LPS-induced RAW264.7 macrophage cells and peritoneal macrophage cells.
MATERIALS AND METHODS
MaterialsDulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), penicillin and streptomycin were obtained from Life Technologies Inc. (Grand Island, NY, U.S.A.). Antibodies against inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), p65, poly(ADP-ribose) polymerase 1 (PARP-1), and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). Horse-radish peroxidase (HRP) conjugated anti-mouse, anti-rabbit and anti-goat immunoglobulin (Ig) were purchased from Jackson Immunoresearch (West Grove, PA, U.S.A.). The enzyme immunoassay (EIA) kits for PGE2, TNF-α, IL-1β and IL-6 were obtained from R&D Systems (Minneapolis, MN, U.S.A.). Random oligonucleotide primers and M-MLV reverse transcriptase were purchased from Promega (Madison, WI, U.S.A.). SYBR green ex Taq was obtained from TaKaRa (Shiga, Japan). iNOS, COX-2, TNF-α, IL-6, IL-1β and β-actin oligonucleotide primers were purchased from Bioneer (Seoul, Korea). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), sulphanilamide, aprotinin, leupeptin, phenylmethylsulfonyl fluoride (PMSF), dithiothreitol (DTT), L-N6-(1-iminoethyl)lysine (L-NIL), N-[2-(cyclohexyloxy)-4-nitrophenyl]methane sulfonamide (NS-398), LPS (Escherichia coli, serotype 0111:B4) and all other chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.).
Plant MaterialThe roots of Achyranthes japonica NAKAI (Amaranthaceae) were obtained from a domestic Korean market (Kyungdong Crude Drugs Market, Seoul, South Korea), in May 2014. The origin of the herbal material was identified by Prof. Dae Sik Jang and a voucher specimen (ACJA-2014) has been deposited in the Lab. of Natural Product Medicine, College of Pharmacy, Kyung Hee University, Republic of Korea.
Extraction and IsolationThe dried and milled plant material (800 g) was extracted with 4 L of 80% EtOH three times by maceration. The extracts were combined and concentrated in vacuo at 40°C to give an 80% EtOH extract (72 g). The 80% EtOH extract (70 g) was chromatographed over Diaion-HP 20 (4.8×50 cm) as stationary phase with H2O–MeOH gradient as mobile phase to afford 18 pooled fractions (E01–E17). Fraction E14 (2.1 g) was subjected to a silica gel column (3.6×25.1 cm, 230–400 mesh) with CH2Cl2–MeOH–H2O mixture (1.0 L of CH2Cl2–MeOH–H2O=8 : 2 : 0.2) to isolate CS (25.3 mg, 0.0032%) and CS-ME (34.6 mg, 0.0043%). The purity of the CS and CS-ME (≥95%) was determined by LC-MS (Supplementary Fig. 1). The chemical structures of CS and CS-ME were identified by spectroscopic data analysis and by comparison with published values.14,15) The analytical results of NMR spectrum were presented as Supplementary Figs. 2–5.
Cell Culture and Sample TreatmentRAW264.7 cells, murine macrophages, were obtained from the Korea Cell Line Bank. These cells were maintained at subconfluence in 95% air and 5% CO2 humidified atmosphere at 37°C. The medium used for routine subculture was DMEM supplemented with 10% FBS, penicillin (100 units/mL) and streptomycin (100 µg/mL). Cells were counted with a hemocytometer and the number of viable cells was determined by trypan blue dye exclusion. Mouse peritoneal macrophages were obtained 4 d after intraperitoneal injection of 2 mL of thioglycollate (4%) to the 10-week-old C57BL/6 male mice and isolated as reported previously.14) Isolated cells form peritoneal cavity were washed twice, resuspended in DMEM, and seeded in culture plates. Cells were incubated with various concentrations of CS, CS-ME, or positive chemicals and then stimulated with LPS (10 ng/mL or 10 µg/mL) for the indicated times.
MTT AssayCells were incubated with a MTT solution for 4 h at 37°C under 5% CO2. One hundred microliters of dimethyl sulfoxide was added to extract the MTT formazan, and the absorbance of each well at 540 nm was read by an automatic microplate reader (Molecular Devices, Sunnyvale, CA, U.S.A.).
Nitrite DeterminationCells were cultured in 24-well plates and then incubated with or without LPS (10 ng/mL; RAW264.7 macrophages or 10 µg/mL; peritoneal macrophages) in the absence or presence of various concentrations of CS or CS-ME for 24 h. Nitrite and nitrate levels in culture media were determined using the Griess reaction.16) Absorbance was then measured at 540 nm using a microplate reader. Fresh culture media were used as blanks in all experiments.
PGE2, TNF-α, IL-6 and IL-1β AssayLevels of PGE2, TNF-α, IL-6, IL-1β in the culture media were quantified using EIA kits, according to the manufacturer’s instructions (R&D Systems).
Protein Extraction and Western Blot AnalysisCells or liver tissues were resuspended in PRO-PREP™ protein extraction solution (Intron Biotechnology, Seoul, Korea) and incubated with 20 min at 4°C. Cell debris was removed by microcentrifugation, followed by quick freezing of the supernatants. The protein concentration was determined using the Bio-Rad protein assay reagent according to the manufacturer’s instruction. Cellular proteins were electroblotted onto a polyvinylidene difluoride (PVDF) membrane following separation on 8–12% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). The immunoblot was incubated with blocking solution (5% skim milk) for 1 h at room temperature, followed by incubation overnight with a primary antibody at 4°C. Blots were washed four times with Tween 20/Tris-buffered saline (T/TBS) and incubated with a 1 : 2000 dilution of HRP-conjugated secondary antibody for 2 h at room temperature. Blots were again washed three times with T/TBS, and then developed by enhanced chemiluminescence (GE Healthcare, WI, U.S.A.). Bio-Rad Quantity One® Software was used for the densitometric analysis.
RNA Isolation and Quantitative Real-Time Reverse-Transcriptase Polymerase Chain Reaction (qRT-PCR)Total cellular RNA was isolated by Easy Blue kits (Intron Biotechnology, Seoul, Korea). From each sample, 1 µg of RNA was reverse-transcribed by MuLV reverse transcriptase, 1 mM deoxyribonucleotide triphosphate (dNTP) and (dT12–18) 0.5 µg/µL. PCR amplification was performed using the incorporation of SYBR green. The oligonucleotide primers for iNOS designed from mouse were CAT GCT ACT GGA GGT GGG TG (forward) and CAT TGA TCT CCG TGA CAG CC (reverse), for COX-2 designed from mouse were GGA GAG ACT ATC AAG ATA GT (forward) and ATG GTC AGT AGA CTT TTA CA (reverse), for IL-1β designed from mouse were TGC AGA GTT CCC CAA CTG GTA CAT C (forward) and GTG CTG CCT AAT GTC CCC TTG AAT C (reverse), for IL-6 were GAG GAT ACC ACT CCC AAC AGA CC (forward) and AAG TGC ATC ATC GTT GTT CAT ACA (reverse), for TNF-α designed from mouse were AGC ACA GAA AGC ATG ATC CG (forward) and CTG ATG AGA GGG AGG CCA TT (reverse). The oligonucleotide primers for β-actin used as a house-keeping gene designed from mouse were ATC ACT ATT GGC AAC GAG CG (forward) and TCA GCA ATG CCT GGG TAC AT (reverse). Steady-state mRNA levels of iNOS, COX-2, IL-1β, IL-6, TNF-α, and β-actin were determined by qPCR using the TaKaRa thermal cycler dice (TaKaRa Bio Inc., Shiga, Japan). A dissociation curve analysis of target mRNAs showed a single peak for each. The mean Ct of the gene of interest was calculated from triplicate measurements and normalized with the mean Ct of a control gene, β-actin.
Transient Transfection and Luciferase AssayRAW264.7 macrophages were co-transfected with pAP-1-Luc or pNF-κB-Luc (Clontech) plasmid plus the phRL-TK plasmid (Promega) using Lipofectamine LTX™ (Invitrogen). Luciferase activity was determined using the Promega luciferase assay system.17)
Nuclear ExtractionNuclear extracts were prepared as described previously with slight modifications. RAW264.7 macrophages cells were pretreated with/without CS-ME (3, 15 or 30 µM), and then stimulated with LPS (10 ng/mL) for 1 h, washed once with phosphate buffered saline (PBS), scraped into 1 mL of cold PBS, and pelleted by centrifugation. Cell pellets were resuspended in hypotonic buffer (10 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2 mM PMSF, 0.5 mM DTT, 10 µg/mL aprotinin) and incubated on ice for 15 min. Cells were then lysed by adding 0.1% Nonidet P-40 and vortexed vigorously for 10 s. Nuclei were pelleted by centrifugation at 12000×g for 1 min at 4°C and resuspended in high salt buffer (20 mM HEPES, pH 7.9, 25% glycerol, 400 mM KCl, 1.5 mM MgCl2, 0.2 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM DTT, 1 mM NaF, 1 mM sodium orthovanadate).
Statistical AnalysisResults are expressed as the mean±standard deviations (S.D.s) of triplicate experiments. Statistically significant values were compared using one-way ANOVA and Dunnett’s post-hoc test, and p values of less than 0.05 were considered statistically significant.
RESULTS AND DISCUSSION
CS-ME Inhibited LPS-Induced NO and PGE2 Production by the Suppression of iNOS and COX-2 Expressions in RAW264.7 MacrophagesThe prototypic endotoxin LPS can potently activate macrophages and induce various proinflammatory mediators.18) Therefore, reducing activation signals in activated macrophages has been suggested as a therapeutic strategy in various inflammatory diseases.18) iNOS and COX-2 are important enzymes that regulate inflammatory processes. iNOS produces an amount of NO, which is responsible for the toxicity of activated macrophages during inflammatory conditions.19,20) COX-2 is also an inducible enzyme that produces PGs during inflammation, and accumulating evidence indicates that PGE2, which is one of the most abundant PGs, is involved in the pain, edema, and vessel permeability associated with inflammatory diseases.21) To investigate whether CS and CS-ME have anti-inflammatory activities, NO and PGE2 production was determined in the presence of CS and CS-ME (30 µM) in LPS-induced RAW264.7 macrophages. CS-ME had a markedly higher inhibitory effects on LPS-induced NO and PGE2 production than CS at 30 µM (Table 1) and concentration-dependently decreased them (Figs. 2A, B). In addition, these inhibitory effects of CS or CS-ME were not caused by nonspecific cytotoxicity, because CS or CS-ME had no effect on cell viability up to 30 µM as determined by MTT assay (Table 1 or Fig. 2A). In general, the pharmacological effects are highly dependent on the polarity of the chemicals because hydrophobic compounds are more permeable to the cell membrane than polar compounds.22) Based on these observations, we suggest that CS-ME is more hydrophobic and likely to be pharmacologically more effective than CS. Previously, it was reported that CS-ME has cytotoxic effects at 25 µM on HCT-116 human colon cancer cells.10) These different cytotoxic results can be caused by the different origin of cells. These cells may have some different sensitivity to the same compound due to different cell membrane features (receptor distribution and membrane permeability) and different genetic features. Next, we investigated whether inhibitory effects of CS-ME on the NO and PGE2 production are from reductions in expressions of iNOS and COX-2. In response to LPS, iNOS and COX-2 at protein and mRNA levels were markedly increased, but CS-ME significantly and concentration-dependently inhibited these up-regulations (Figs. 2C, E). To determine whether CS-ME effects on iNOS and COX-2 mRNA stability, cells were pretreated with LPS for 12 h and then treated with actinomycin D (an inhibitor of gene transcription). CS-ME did not affect mRNA stability of iNOS and COX-2 in LPS-induced RAW264.7 macrophages (Supplementary Fig. 6). Thus, observed inhibitions of the production of NO and PGE2 may be attributed to the suppressions of the transcriptions of iNOS and COX-2 mRNA and subsequent reductions in their protein expressions.
Table 1. Effects of CS and CS-ME on LPS-Induced NO and PGE
2 Production and Cell Viability in RAW264.7 Macrophages
| Sample | Inhibition (%, 30 µM) | Cell viabilityc) (%, 30 µM) |
|---|
| NOa) | PGE2b) |
|---|
| CS | 46.47±6.34 | 0 | 105±5.10 |
| CS-ME | 74.80±6.12 | 33.36±7.18 | 98±2.20 |
a) Levels of NO in the culture media was quantified using Griess reaction. b) Levels of PGE2 in the culture media was quantified using an EIA. c) Cell viability was determined using a MTT assay.
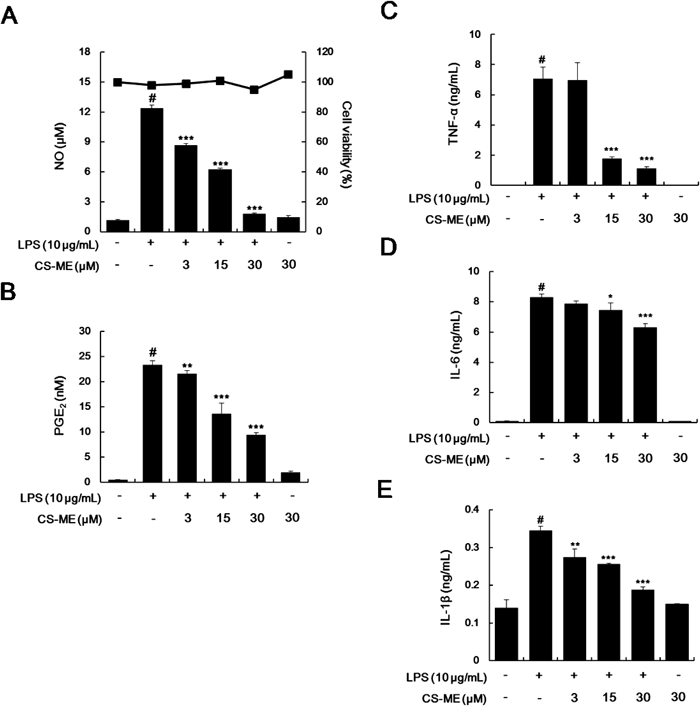
The anti-inflammatory effects of CS-ME are supported by the results of our investigations of its effects on several pro-inflammatory cytokines, namely, TNF-α, IL-6, and IL-1β in LPS-induced macrophages, because they are known to have profound effects on the regulation of immune reactions, hematopoiesis, inflammation, and, in some cases, shock and death.23) We investigated the effects of CS-ME on LPS-induced TNF-α, IL-6 and IL-1β production and their mRNA levels using EIAs and qRT-RCR, respectively. In resting cells, TNF-α, IL-6 and IL-1β levels were low, but in response to LPS, these levels obviously increased. Pretreatment with CS-ME significantly reduced LPS-induced production of TNF-α, IL-6 and IL-1β (Figs. 3A, C). CS-ME also attenuated the LPS-induced TNF-α, IL-6 and IL-1β mRNA expressions (Figs. 3D, F). Inhibitory effect of CS-ME on the TNF-α production is much more potent than that on its mRNA expression. So, we measured the effects of CS-ME on TNF-α mRNA stability in order to examine the post-transcriptional regulatory effects of CS-ME. However, it has been shown that TNF-α mRNA stability showed no difference in the presence or absence of CS-ME by mRNA decay experiments (Supplementary Fig. 6). Since production of TNF-α goes through a lot of translational modification step after transcription, further studies are necessary to examine whether CS-ME could have influence on these process. Collectively, these results indicate that CS-ME could have anti-inflammatory activities via inhibition of the expressions of iNOS, COX-2 and other pro-inflammatory genes.
CS-ME Inhibited LPS-Induced Production of NO, PGE2, and Pro-inflammatory Cytokines in Mouse Peritoneal MacrophagesTo determine whether the anti-inflammatory effects of CS-ME observed in RAW264.7 macrophage cells also occur in primary cells, we examined the effect of CS-ME on the LPS-induced production of NO, PGE2, TNF-α, IL-6 and IL-1β in peritoneal macrophages isolated from C57BL/6 mice. In these cells, CS-ME was found to inhibit the LPS-induced production of NO, PGE2, and pro-inflammatory cytokines significantly and concentration-dependently (Figs. 4A, E). We confirmed that these inhibitory effects of CS-ME were not caused by a cytotoxic effect (determined by MTT assay, Fig. 4A).
CS-ME Inhibited LPS-Induced NF-κB and AP-1 Activation in RAW264.7 MacrophagesTranscription factors, such as NF-κB and AP-1 have been known to regulate the expressions of inflammatory mediators including iNOS, COX-2, TNF-α, and IL-6.24) We examined the effect of CS-ME on LPS-induced NF-κB- and AP-1-dependent reporter gene activities. Analysis of reporter gene expression using pNF-κB-luc and pAP-1-luc demonstrated that CS-ME concentration-dependently inhibited luciferase activity (Figs. 5A, C). Based on these results, we suggest that the inhibition of the expressions of pro-inflammatory mediators by CS-ME might be caused by the suppression of NF-κB and AP-1 activity. To identify the mechanisms involved in the inhibition of NF-κB and AP-1 activity by CS-ME, we tested the effect of CS-ME on NF-κB and AP-1 activation signals. It is commonly known that NF-κB consisting of p50/p65 heterodimer binds to inhibitory κBs (IκBs) molecules via nuclear localization sequence of p65 subunit and present in the cytoplasm under normal conditions. Once activated by LPS, NF-κB escapes from IκB and translocate to the nucleus, where it binds to its consensus DNA binding site to induce the the transcription of pro-inflammatory genes.25) Accordingly, we investigated whether CS-ME prevents the nuclear translocation of the p65 subunit of NF-κB. It was found that pretreatment with CS-ME attenuated the LPS-induced nuclear translocation of p65 at indicated times (Fig. 5B). AP-1 activity is regulated at two major levels, that is, its activity is increased due to the expression and phosphorylation of c-Fos and c-Jun by mitogen-activated protein kinase (MAPK).26) LPS increases both the expression and phosphorylation of c-Fos and c-Jun in macrophages.27,28) Therefore, we examined the effect of CS-ME on the expression of c-Fos and c-Jun in cytosol fraction and found CS-ME showed an inhibitory effect on the expression of c-Fos, whereas it did not influence on c-Jun (Fig. 5D). Moreover, CS-ME inhibited LPS-induced phosphorylation of c-Fos instead of c-Jun in nuclear fraction (Fig. 5E). These results suggested that the inhibitory effects of CS-ME on AP-1 transcriptional activity could be caused by the reduction of expression and decreased phosphorylation of c-Fos, but not by the nuclear translocation. In addition, it was found that CS-ME did not show any inhibitory effects on the c-Jun expression, phosphorylation and translocation. Recently, it was reported that chikusetsusaponin V has anti-inflammatory activities in RAW264.7 macrophages.29) Chikusetsusaponin V and CS-ME are dammarane-type triterpenoids and are different only in glycosidic compositions. Similar to the CS-ME, chikusetsusaponin V inhibited LPS-induced pro-inflammatory mediators (NO, TNF-α, IL-1β) via NF-κB signaling inactivation. Chikusetsusaponin V inhibited the phosphorylation of MAPK (Jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK)) and mRNA expressions of TLR4 and CD14. From these results, Wang et al. suggested that chikusetsusaponin V could regulate NF-κB and MAPK signaling pathways by inhibiting the binding of LPS to TLR4 on macrophages.29) However, they did not test the effects of chikusetsusaponin V on AP-1 activation. Since it has been well-known that MAPK activities strongly regulate the AP-1 activation via their phosphorylation,30) anti-inflammatory activity signaling of chikusetsusaponin V and CS-ME could be similar in LPS-induced RAW264.7 macrophages. However, an in-depth and in vivo animal studies are needed to unravel the anti-inflammatory molecular mechanisms of CS-ME.
In summary, the present study suggested that CS-ME, a triterpenoid saponin isolated from the dried roots of A. japonica, attenuates the LPS-induced production of NO, PGE2, TNF-α, IL-6, and IL-1β in macrophages. Molecular data revealed that the inhibitory effects on iNOS, COX-2, TNF-α, IL-6, and IL-1β expressions of CS-ME were involved in inactivation of NF-κB and AP-1 transcription factor. Further studies are needed to examine the anti-inflammatory effects of CS-ME in an animal model.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), which is funded by the Ministry of Education, Science and Technology (No. 2013R1A1A2011043).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Rankin JA. Biological mediators of acute inflammation. AACN Clin. Issues, 15, 3–17 (2004).
- 2) Guzik TJ, Korbut R, Adamek-Guzik T. Nitric oxide and superoxide in inflammation and immune regulation. J. Physiol. Pharmacol., 54, 469–487 (2003).
- 3) Medzhitov R, Janeway CA Jr. Innate immunity: the virtues of a nonclonal system of recognition. Cell, 91, 295–298 (1997).
- 4) Shin JS, Park YM, Choi JH, Park HJ, Shin MC, Lee YS, Lee KT. Sulfuretin isolated from heartwood of Rhus verniciflua inhibits LPS-induced inducible nitric oxide synthase, cyclooxygenase-2, and pro-inflammatory cytokines expression via the down-regulation of NF-kappaB in RAW264.7 murine macrophage cells. Int. Immunopharmacol., 10, 943–950 (2010).
- 5) Barnes PJ, Karin M. Nuclear factor-kappaB: a pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med., 336, 1066–1071 (1997).
- 6) Khalaf H, Jass J, Olsson PE. Differential cytokine regulation by NF-kappaB and AP-1 in Jurkat T-cells. BMC Immunol., 11, 26 (2010).
- 7) Youn GS, Kwon DJ, Ju SM, Rhim H, Bae YS, Choi SY, Park J. Celastrol ameliorates HIV-1 Tat-induced inflammatory responses via NF-kappaB and AP-1 inhibition and heme oxygenase-1 induction in astrocytes. Toxicol. Appl. Pharmacol., 280, 42–52 (2014).
- 8) Bang SY, Kim J-H, Kim H-Y, Lee YJ, Park SY, Lee SJ, Kim Y. Achyranthes japonica exhibits anti-inflammatory effect via NF-kappaB suppression and HO-1 induction in macrophages. J. Ethnopharmacol., 144, 109–117 (2012).
- 9) Park JH, Kang SN, Shin D, Hur IC, Kim IS, Jin SK. Antioxidant activities of Achyranthes japonica Nakai extract and its application to the pork sausages. Asian-australas. J. Anim. Sci., 26, 287–294 (2013).
- 10) Lee KM, Yun JH, Lee DH, Park YG, Son KH, Nho CW, Kim YS. Chikusetsusaponin IVa methyl ester induces cell cycle arrest by the inhibition of nuclear translocation of beta-catenin in HCT116 cells. Biochem. Biophys. Res. Commun., 459, 591–596 (2015).
- 11) Cui J, Xi MM, Li YW, Duan JL, Wang L, Weng Y, Jia N, Cao SS, Li RL, Wang C, Zhao C, Wu Y, Wen AD. Insulinotropic effect of Chikusetsu saponin IVa in diabetic rats and pancreatic beta-cells. J. Ethnopharmacol., 164, 334–339 (2015).
- 12) Sadzak I, Schiff M, Gattermeier I, Glinitzer R, Sauer I, Saalmuller A, Yang E, Schaljo B, Kovarik P. Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl. Acad. Sci. U.S.A., 105, 8944–8949 (2008).
- 13) Dahmer T, Berger M, Barlette AG, Reck J Jr, Segalin J, Verza S, Ortega GG, Gnoatto SC, Guimaraes JA, Verli H, Gosmann G. Antithrombotic effect of chikusetsusaponin IVa isolated from Ilex paraguariensis (Mate). J. Med. Food, 15, 1073–1080 (2012).
- 14) Alleva DG, Johnson EB, Lio FM, Boehme SA, Conlon PJ, Crowe PD. Regulation of murine macrophage proinflammatory and anti-inflammatory cytokines by ligands for peroxisome proliferator-activated receptor-gamma: counter-regulatory activity by IFN-gamma. J. Leukoc. Biol., 71, 677–685 (2002).
- 15) Sakai S, Katsumata M, Satoh Y, Nagasao M, Miyakoshi M, Ida Y, Shoji J. Oleanolic acid saponins from root bark of Aralia elata. Phytochemistry, 35, 1319–1324 (1994).
- 16) Miranda KM, Espey MG, Wink DA. A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide, 5, 62–71 (2001).
- 17) Shin JS, Yun KJ, Chung KS, Seo KH, Park HJ, Cho YW, Baek NI, Jang D, Lee KT. Monotropein isolated from the roots of Morinda officinalis ameliorates proinflammatory mediators in RAW264.7 macrophages and dextran sulfate sodium (DSS)-induced colitis via NF-kappaB inactivation. Food Chem. Toxicol., 53, 263–271 (2013).
- 18) Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front. Biosci., 13, 453–461 (2008).
- 19) Gross SS, Wolin MS. Nitric oxide: pathophysiological mechanisms. Annu. Rev. Physiol., 57, 737–769 (1995).
- 20) MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu. Rev. Immunol., 15, 323–350 (1997).
- 21) Morteau O. Prostaglandins and inflammation: the cyclooxygenase controversy. Arch. Immunol. Ther. Exp., 48, 473–480 (2000).
- 22) Levitan H, Barker JL. Salicylate: a structure–activity study of its effects on membrane permeability. Science, 176, 1423–1425 (1972).
- 23) Christiaens I, Zaragoza DB, Guilbert L, Robertson SA, Mitchell BF, Olson DM. Inflammatory processes in preterm and term parturition. J. Reprod. Immunol., 79, 50–57 (2008).
- 24) Tak PP, Firestein GS. NF-kappaB: a key role in inflammatory diseases. J. Clin. Invest., 107, 7–11 (2001).
- 25) Hoesel B, Schmid JA. The complexity of NF-kappaB signaling in inflammation and cancer. Mol. Cancer, 12, 86 (2013).
- 26) Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem., 270, 16483–16486 (1995).
- 27) Suh HW, Choi SS, Lee JK, Lee HK, Han EJ, Lee J. Regulation of c-fos and c-jun gene expression by lipopolysaccharide and cytokines in primary cultured astrocytes: effect of PKA and PKC pathways. Arch. Pharm. Res., 27, 396–401 (2004).
- 28) Hambleton J, Weinstein SL, Lem L, DeFranco AL. Activation of c-Jun N-terminal kinase in bacterial lipopolysaccharide-stimulated macrophages. Proc. Natl. Acad. Sci. U.S.A., 93, 2774–2778 (1996).
- 29) Wang H, Qi J, Li L, Wu T, Wang Y, Wang X, Ning Q. Inhibitory effects of Chikusetsusaponin IVa on lipopolysaccharide-induced pro-inflammatory responses in THP-1 cells. Int. J. Immunopathol. Pharmacol., 28, 308–317 (2015).
- 30) Karin M, Hawkins PT. The regulation of AP-1 activity by mitogen-activated protein kinases. Philos. Trans. R. Soc. Lond. B Biol. Sci., 351, 127–134 (1996).