Abstract
Anchorage-independent growth is one of the defining characteristics of cancer cells. Many oncogenes and tumor suppressor genes are involved in regulating this type of growth. Factor for adipocyte differentiation 104 gene (fad104) is a regulator of adipogenesis and osteogenesis. Previously, we reported that fad104 suppressed metastasis as well as invasion of melanoma cells. However, it is unclear whether fad104 is involved in malignant transformation, which is associated with metastasis. In this study, we revealed that fad104 negatively regulated the colony forming activity of melanoma cells. The presence of the N-terminal region of FAD104 was required for the regulation of malignant transformation of melanoma cells. In addition, the deletion mutant of FAD104 that contained the N-terminal region and transmembrane domain interacted with signal transducer and activator of transcription 3 (STAT3) and suppressed STAT3 activity. However, the deletion mutant of FAD104 lacking the N-terminal region did not influence the interaction with STAT3 or suppress the STAT3 activity. Moreover, FAD104 interacted with the C-terminal region of STAT3. In summary, we demonstrated that fad104 suppressed anchorage-independent growth of melanoma cells, and that the N-terminal region of FAD104 is essential for inhibiting malignant transformation and STAT3 activity.
Melanoma is one of the most frequently occurring malignant tumors and is characterized by its high rate of invasion and metastasis. The development of metastatic diseases is highly complex. Anchorage-independent growth is a crucial step in the acquisition of metastatic potential.1) Tumor cells that have a high potential for anchorage-independent growth possess the ability to migrate through the body, colonize other tissues, and grow metastatically.2) Therefore, understanding the mechanisms by which anchorage-independent cancer cell growth is regulated is essential for the development of therapies for metastatic cancer. Although multiple genetic factors for anchorage-independent growth have been identified, little is known about the molecular basis for the capacity for anchorage-independent growth.3,4)
Previously, using the polymerase chain reaction (PCR) subtraction method, we identified unknown genes, whose expressions were elevated at the early period in adipogenesis, such as factor for adipocyte differentiation 24 gene (fad24), fad49, fad104, and fad158, and showed that they promoted adipocyte differentiation in mouse 3T3-L1 cells.5–10) The expression of fad104, also known as fndc3b, is transiently increased 3 h after adipogenic induction. FAD104 has a proline-rich region, nine fibronectin type III domains, and a transmembrane region. Our previous study revealed that fad104 functioned as positive and negative regulator for adipogenesis and osteoblast differentiation, respectively.11) Furthermore, we showed that fad104 is essential for lung maturation.12,13)
Recently, we also reported that fad104 suppressed the invasion and metastasis of melanoma cells by inhibiting signal transducer and activator of transcription 3 (STAT3) activity.14) However, it remains unknown whether fad104 is involved in anchorage-independent growth that appears to be closely associated with metastasis. In this paper, we revealed that fad104 inhibited the colony formation ability of melanoma cells. Moreover, the effect is eliminated in melanoma cells over-expressing fad104 lacking the N-terminal region. Furthermore, FAD104 interacts with the C-terminal region of STAT3. In addition, the deletion mutant of fad104 lacking fibronectin type III domain, but having the N-terminal region and transmembrane domain, suppressed the phosphorylation and transcriptional activity of STAT3, but the deletion mutant of fad104 lacking the N-terminal region did not do so. These findings indicate that the presence of the N-terminal region of fad104 is crucial for suppression of malignant transformation and STAT3 activity in melanoma cells.
MATERIALS AND METHODS
Plasmid Constructionp3xFLAG-fad104FL, a FLAG-tagged FAD104 expression plasmid, and p3xFLAG-fad104ΔN, a FLAG-tagged FAD104 mutant expression plasmid lacking the N-terminal proline rich motif (Δ1–212), have been previously described.15) p3xFLAG-tagged fad104ΔFNIII, a FLAG-tagged FAD104 mutant expression plasmid lacking the central nine FNIII domains, was constructed by subcloning the fragment amplified by PCR. The expression plasmid of full length and a series of deletion mutants of STAT3 were generated by PCR using primers including HindIII and SalI sites. The PCR products were subcloned into p3xFLAG-CMV™-7.1 (Sigma, MO, U.S.A.).
Cell Culture and Interleukin 6 (IL-6) StimulationA375SM and B16F10 cells were cultured as previously described.14) For IL-6 stimulation, A375SM cells were treated with a medium containing 5 ng/mL of IL-6 for 30 min.
Generation of Stable Cell LinesGeneration of B16F10 cells stably expressing FAD104FL or ΔN was according to previous publication.14) In brief, B16F10 cells were transfected with p3xFLAG-fad104FL or ΔN, and selected for neomycin resistance. B16F10 cells introduced with empty vector were used as a control.
Soft Agarose AssayB16F10 cells (2.5×103) stably over-expressing FAD104FL or ΔN were cultured in 0.35% soft agarose (10% fetal bovine serum (FBS)-Dulbecco’s modified Eagle’s medium (DMEM)) layered on a 0.5% hard agarose (10% FBS-DMEM) in 35-mm dish. After 15 d, colonies were stained with 0.01% crystal violet for counting.
Immunofluorescence StainingFLAG-tagged FAD104 expression plasmids were transfected into A375SM cells plated onto cell disks (SUMITOMO BAKELITE, Tokyo, Japan) using polyethylenimine (PEI) one day before transfection. Cells were fixed with 4% paraformaldehyde (PFA) and incubated with mouse monoclonal anti-DYKDDDDK tag antibody for the detection of FLAG tag (Wako Pure Chemical Industries, Ltd., Osaka, Japan) for 1 h at room temperature. After washing the disk three times, tetramethylrhodamine (TRITC)-conjugated goat anti-mouse immunoglobulin G (IgG) (Sigma) was added and incubated for 30 min at room temperature. The signals for TRITC were detected by fluorescence microscopy (BX51; Olympus, Tokyo, Japan).
Analysis of Morphological ChangeUsing PEI, FLAG-tagged full length or deletion mutants of FAD104 expression plasmids were introduced into A375SM cells. Cells were fixed with 4% PFA and incubated with anti-DYKDDDDK antibody described above. After washing, fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG (Sigma) was added and incubated for 30 min at room temperature. After washing three times, TRITC-labeled phalloidin (Sigma) was incubated for 30 min at room temperature. The signals for FITC and TRITC were detected by fluorescence microscopy (BX51; Olympus).
ImmunoprecipitationEither the p3xFLAG-fad104FL or p3xFLAG-fad104ΔN plasmid was transfected into A375SM cells using PEI. Immunoprecipitation protocol was described earlier.14) The bound proteins were separated, and immunoblotted with anti-DDDDK tag antibody for the detection of FLAG tag (MBL, Nagoya, Japan).
For detection of interactions between FAD104ΔFNIII and STAT3, A375SM cells were transfected with the p3xFLAG-fad104ΔFNIII plasmid. After 24 h, cell lysates were prepared with Nonidet-P40 lysis buffer, and were incubated with 3.8 µg anti-FLAG antibody (Sigma) overnight at 4°C. The mixtures were rotated with protein G-coupled Sepharose beads for 4 h at 4°C. For a negative control experiment, normal mouse IgG was used instead of anti-FLAG antibody. The bound proteins were detected by Western blotting using anti-STAT3 antibody (Santa Cruz, CA, U.S.A.).
Glutathione S-Transferase (GST) Pull-Down AssayExpression of GST and GST-FAD104N and GST pull-down assay were performed as described previously.14) GST alone or GST-FAD104 fusion protein bound beads were incubated with equal amounts of cell lysates prepared from A375SM cells, which were transfected with either full length or deletion mutants of STAT3 expression plasmid, and subsequently rotated overnight at 4°C. After washing, the bound proteins were detected by Western blotting.
Western BlottingPreparation of cell lysates and Western blot analyses were according to previous publication.14) Primary antibodies for FAD104 (1 : 400), phospho-STAT3 (Y705) (1 : 1000; Cell Signaling, MA, U.S.A.), STAT3 (1 : 1000; Santa Cruz), DDDDK tag for detection of FLAG tag (1 : 2000; MBL), and β-actin (1 : 100000; Sigma) were used. A polyclonal FAD104 antibody against the N-terminal region of mouse FAD104 (PHKKLKDRQIDR, amino acids 191–202) was prepared in our laboratory.13) The band intensity of the blots was quantified with Image-J software.
Luciferase AssayTwenty-five nanograms of p4xM67-tk-Luc as a reporter gene was transfected with 75 ng of FLAG-tagged FAD104FL, ΔN, ΔFNIII expression plasmid, or FLAG-empty plasmid into A375SM cells, which were seeded in 24 well plates one day before transfection. Twenty-four hours after transfection, the cell lysates were prepared and luciferase assay was performed. The transfection efficiency was normalized with the β-galactosidase activity, derived from co-transfected plasmid cytomegalovirus (pCMV)-β-gal (6.25 ng).
Statistical TestsStatistics were analysed using R (http://cran.r-project.org/), and one-way ANOVA with post-hoc Tukey–Kramer honestly significant difference (HSD) test.
RESULTS
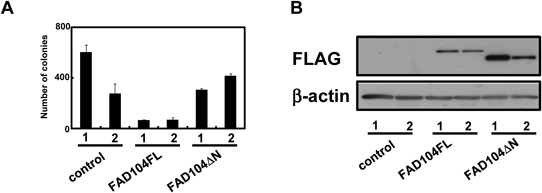
FAD104 Inhibits the Colony Formation Ability of B16F10 Cells via the Presence of the N-Terminal Region of FAD104To reveal whether fad104 regulates colony-forming activity, we verified the effect of over-expression of FAD104 on anchorage-independent growth of B16F10 cells. Stable transformants of B16F10 expressing FAD104FL revealed a remarkable decrease in colony-forming ability compared with control B16F10 cells in soft agarose assay (Fig. 1A). Previously, we have demonstrated that the N-terminal region of FAD104 interacted with STAT3 by using a GST pull down assay.14) Therefore, we focused on the N-terminal region of FAD104 and made another stable transformant expressing FAD104ΔN. The colony numbers of two groups of FAD104ΔN over-expressing cells did not differ from those of control cells (Fig. 1A), although the protein expression levels of FAD104ΔN was slightly higher than those of FAD104FL (Fig. 1B). These results strongly indicate that the over-expression of fad104 suppresses the ability of anchorage-independent growth of B16F10 cells via the presence of the N-terminal region of FAD104.
N-Terminal Region of FAD104 Is Essential for Interaction with STAT3Our previous study revealed the interaction of FAD104 with STAT3 under physiological conditions.14) To further characterize this binding, we generated a FLAG-FAD104ΔFNIII, only having the proline-rich N-terminal region and transmembrane domain of FAD104, in addition to the FLAG-tagged FAD104ΔN expression plasmid (Fig. 2A). We first investigated the localization of these deletion mutants. Our previous studies have shown that FAD104 localized in the endoplasmic reticulum.12) As shown in Fig. 2B, both FAD104ΔN and FAD104ΔFNIII localized in the endoplasmic reticulum as well as FAD104FL. We next investigated the effect of wild-type and mutated FAD104 on cell morphology. The F-actin staining with phalloidin showed that the cell shapes of FLAG-tagged FAD104FL and two deletion mutants over-expressing cells were indistinguishable from those of non-transfected cells which were non-detectable levels of FLAG signal, indicating that FAD104FL and deletion mutants did not influence the cell morphology (Fig. 2C). Using an immunoprecipitation analyses, we next investigated whether the N-terminal region of FAD104 is required for interaction with STAT3. FAD104FL interacted with an STAT3 but FAD104ΔN did not bind to STAT3 (Fig. 2D). Although we also verified the interaction between FAD104ΔFNIII and STAT3 by using an immunoprecipitation assay with anti-STAT3 antibody, we could not evaluate the interaction due to the detection of nonspecific bands. Therefore, the immunoprecipitation of FAD104ΔFNIII was performed using an anti-FLAG antibody, and the bound STAT3 was detected. As a result, FAD104ΔFNIII was found to interact with STAT3 (Fig. 2E). These results strongly indicate that the N-terminal region of FAD104 is essential to interacting with STAT3.
FAD104 Interacts with C-Terminal of STAT3To characterize the domains responsible for the interaction with FAD104, we generated three deletion mutants of STAT3 as shown in Fig. 3A. A GST pull down assay showed that GST-FAD104N strongly bound to the C-terminal region and slightly interacted with the N-terminal region of STAT3, whereas the linker domain of STAT3 failed to associate with GST-FAD104N (Fig. 3B). These results indicate that FAD104 mainly interacted with the C-terminal region of STAT3 in melanoma cells.
The N-Terminal Region of FAD104 Is Required to Regulate STAT3 ActivityWe next tested whether the N-terminal region of FAD104 is necessary for the regulation of STAT3 activity. It is reported that IL6 triggered a signaling cascade and led to the phosphorylation of STAT3.16) We transfected A375SM cells with either a FAD104FL, FAD104ΔN or FAD104ΔFNIII expression plasmid and treated the cells with 5 ng/mL IL6 for 30 min. The level of phospho-STAT3 increased in control cells after IL6 treatment (Figs. 4A, B). However, that of STAT3 in over-expressing FAD104FL showed significantly low compared with that of control cells treated with IL6. Over-expression of FAD104ΔFNIII also remarkably suppressed the phosphorylation levels of STAT3 as well as FAD104FL, whereas over-expressing FAD104ΔN did not reduce it. We next examined the effect of the presence or absence of the N-terminal region of FAD104 on the transcription ability of STAT3 by transfection analysis using a STAT3 reporter plasmid (4xM67-tk-Luc) containing four copies of binding elements for STAT3. Both of FAD104FL and FAD104ΔFNIII significantly inhibited reporter gene activity (Fig. 4C). In contrast, FAD104ΔN did not affect it. The presence of the N-terminal region of FAD104 appears to be required for the negative regulation of phosphorylation and of ability as a transcription factor of STAT3.
DISCUSSION
In this study, we showed that over-expression of fad104 suppressed anchorage-independent growth of melanoma cells. Because anchorage-independent growth has been connected with tumorigenesis and metastasis in vivo, identifying molecular mechanisms by which anchorage-independent growth is regulated is crucial for understanding aggressive tumors. It is known that multiple factors are involved in the regulation of colony forming activity of cancer cells. In particular, STAT3 is known as a crucial factor for anchorage-independent growth. It is reported that STAT3 promotes anchorage-independent growth of cancer cells and inhibition of STAT3 homodimerization suppresses malignant transformation.17,18) Moreover, STAT3 inhibition suppressed tumor growth of B16F10 cells.19)
While full-length FAD104 inhibited the colony forming activity of melanoma cells, FAD104ΔN did not reduce it (Fig. 1A). Furthermore, we also showed that the N-terminal region of FAD104 is required for the suppression of STAT3 activity (Fig. 4), suggesting that fad104 suppresses anchorage-independent growth through the inhibition of STAT3 activity; the presence of the N-terminal region of FAD104 appears to be required for its function. Although we indicated that fad104 repressed malignant transformation, it is still unknown whether fad104 is involved in tumorigenicity. To further characterize the function of fad104 on tumor malignancy, it is necessary to investigate the effects of fad104 on tumor formation in vivo using a xenograft mouse model. Furthermore, it has been reported that over-expression of the constitutively activated STAT3 mutant transfected into NIH3T3 fibroblast induced cellular transformation and tumor formation in nude mice.20,21) It is necessary to validate whether fad104 is involved in the transformation of normal cells.
The C-terminal region of the STAT3 encoding SH2 domain is required for the phosphorylation of STAT3. The phosphorylation site of STAT3 (Y705) exists in the transactivation domain. It is reported that STAT3 binds to respective tyrosine residues on gp130, a common receptor of IL6 superfamily, through its SH2 domain and is subsequently phosphorylated on a single tyrosine residue (Y705) at the C-terminal region by the Janus kinases (JAKs).22–24) In this paper, we revealed that FAD104 interacts with the C-terminal region of STAT3 (Fig. 3). Moreover, we also clarified that fad104 decreased the phosphorylation level of STAT3 via the N-terminal region of FAD104. These results suggest that the N-terminal region of FAD104 masks the STAT3 phosphorylation site and inhibits binding between STAT3 and gp130, resulting in the reduction of phosphorylation of STAT3. In addition, the activation of STAT3 is ingeniously regulated by other factors, such as protein tyrosine phosphatases, inhibitory factors, and factors for negative feedback.25–27) Therefore, an investigation as to whether fad104 controls activation of STAT3 via cooperation with those should be needed.
FAD104 localizes to endoplasmic reticulum (ER).12) Some reports demonstrated that the protein localized to ER contributes to STAT3 signaling and cancer progression. For example, ERp57, an ER resident thiol disulfide oxidoreductase, suppresses the phosphorylation level of STAT3.28) Additionally, protein kinase R-like ER kinase (PERK), an unfolded protein response (UPR) transducer localized to ER membrane, activates STAT3.29) Moreover, it is reported that calreticulin, a chaperon protein involved in ER stress, promotes invasion and tumorigenesis through the activation of STAT3 signaling.30) These findings provide evidence that there may be important interaction between ER-ER stress-STAT3 signaling and tumor progression. It is needed to investigate the effect of fad104 on the function of these factors involved in ER stress.
Previously, we showed that fad104-deficient mice died at birth because of lung abnormalities and that calvarial bone formation was promoted fad104-deficient mice, suggesting that fad104 played important roles for lung maturation and ossification.13,15) Since STAT3 promotes lung and born formation,31,32) it is possible that fad104 regulates STAT3 signal even in lung maturation and osteoblast differentiation.
Our present and previous findings raise the possibility that fad104 contributes to tumor malignancy. In immunohistochemistry-based protein profiling data available from the Human Protein Atlas (www.proteinatlas.org),33) it is reported that FAD104 expresses in various human tissue such as adipose tissue, pancreas, and kidney. Additionally, the expression levels of FAD104 in most malignant tumors including pancreatic and renal cancer are lower than those of corresponding normal tissues, implying that decline of the expression level of fad104 may be involved in tumor malignancy.
In conclusion, our present findings provide evidence that fad104 negatively regulates anchorage-independent growth of melanoma cells. In addition, we found that the presence of the N-terminal region of FAD104 was required for inhibiting anchorage-independent growth and STAT3 activity. Although further studies are required, this work should contribute to further understanding of the mechanisms involved in tumor malignancy.
Acknowledgments
This work was supported in part by Grants from JSPS KAKENHI (No. 26460074 to MN, and No. 21390024 to MI).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: Correlation with cell growth in semi-solid medium. Cell, 3, 355–359 (1974).
- 2) Gassmann P, Haier J. The tumor cell–host organ interface in the early onset of metastatic organ colonization. Clin. Exp. Metastasis, 25, 171–181 (2008).
- 3) Cifone MA, Fidler IJ. Correlation of patterns of anchorage-independent growth with in vivo behavior of cells from a murine fibrosarcoma. Proc. Natl. Acad. Sci. U.S.A., 77, 1039–1043 (1980).
- 4) Mori S, Chang JT, Andrechek ER, Matsumura N, Baba T, Yao G, Kim JW, Gatza M, Murphy S, Nevins JR. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene, 28, 2796–2805 (2009).
- 5) Imagawa M, Tsuchiya T, Nishihara T. Identification of inducible genes at the early stage of adipocyte differentiation of 3T3-L1 cells. Biochem. Biophys. Res. Commun., 254, 299–305 (1999).
- 6) Nishizuka M, Tsuchiya T, Nishihara T, Imagawa M. Induction of Bach1 and ARA70 gene expression at an early stage of adipocyte differentiation of mouse 3T3-L1 cells. Biochem. J., 361, 629–633 (2002).
- 7) Tominaga K, Johmura Y, Nishizuka M, Imagawa M. Fad24, a mammalian homolog of Noc3p, is a positive regulator in adipocyte differentiation. J. Cell Sci., 117, 6217–6226 (2004).
- 8) Hishida T, Eguchi T, Osada S, Nishizuka M, Imagawa M. A novel gene, fad49, plays a crucial role in the immediate early stage of adipocyte differentiation via involvement in mitotic clonal expansion. FEBS J., 275, 5576–5588 (2008).
- 9) Tominaga K, Kondo C, Johmura Y, Nishizuka M, Imagawa M. The novel gene fad104, containing a fibronectin type III domain, has a significant role in adipogenesis. FEBS Lett., 577, 49–54 (2004).
- 10) Tominaga K, Kondo C, Kagata T, Hishida T, Nishizuka M, Imagawa M. The novel gene fad158, having a transmembrane domain and leucine-rich repeat, stimulates adipocyte differentiation. J. Biol. Chem., 279, 34840–34848 (2004).
- 11) Kishimoto K, Kato A, Osada S, Nishizuka M, Imagawa M. Fad104, a positive regulator of adipogenesis, negatively regulates osteoblast differentiation. Biochem. Biophys. Res. Commun., 397, 187–191 (2010).
- 12) Nishizuka M, Kishimoto K, Kato A, Ikawa M, Okabe M, Sato R, Niida H, Nakanishi M, Osada S, Imagawa M. Disruption of the novel gene fad104 causes rapid postnatal death and attenuation of cell proliferation, adhesion, spreading and migration. Exp. Cell Res., 315, 809–819 (2009).
- 13) Kishimoto K, Nishizuka M, Ueda T, Kajita K, Ugawa S, Shimada S, Osada S, Imagawa M. Indispensable role of factor for adipocyte differentiation 104 (fad104) in lung maturation. Exp. Cell Res., 317, 2110–2123 (2011).
- 14) Katoh D, Nishizuka M, Osada S, Imagawa M. Fad104, a positive regulator of adipocyte differentiation, suppresses invasion and metastasis of melanoma cells by inhibition of STAT3 activity. PLoS ONE, 10, e0117197 (2015).
- 15) Kishimoto K, Nishizuka M, Katoh D, Kato A, Osada S, Imagawa M. FAD104, a regulatory factor of adipogenesis, acts as a novel regulator of calvarial bone formation. J. Biol. Chem., 288, 31772–31783 (2013).
- 16) Heinrich PC, Behrmann I, Müller-Newen G, Schaper F, Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. Biochem. J., 334, 297–314 (1998).
- 17) Tsareva SA, Moriggl R, Corvinus FM, Wiederanders B, Schütz A, Kovacic B, Friedrich K. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia, 9, 279–291 (2007).
- 18) Zhang X, Sun Y, Pireddu R, Yang H, Urlam MK, Lawrence HR, Guida WC, Lawrence NJ, Sebti SM. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res., 73, 1922–1933 (2013).
- 19) Blaskovich MA, Sun J, Cantor A, Turkson J, Jove R, Sebti SM. Discovery of JSI-124 (cucurbitacin I), a selective Janus kinase/signal transducer and activator of transcription 3 signaling pathway inhibitor with potent antitumor activity against human and murine cancer cells in mice. Cancer Res., 63, 1270–1279 (2003).
- 20) Turkson J, Bowman T, Garcia R, Caldenhoven E, De Groot RP, Jove R. Stat3 activation by Src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol., 18, 2545–2552 (1998).
- 21) Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol., 18, 2553–2558 (1998).
- 22) Lütticken C, Wegenka UM, Yuan J, Buschmann J, Schindler C, Ziemiecki A, Harpur AG, Wilks AF, Yasukawa K, Taga T, Kishimoto T, Barbieri G, Pellegrini S, Sendtner M, Heinrich PC, Horn F. Association of transcription factor APRF and protein kinase Jak1 with the interleukin-6 signal transducer gp130. Science, 263, 89–92 (1994).
- 23) Stahl N, Boulton TG, Farruggella T, Ip NY, Davis S, Witthuhn BA, Quelle FW, Silvennoinen O, Barbieri G, Pellegrini S, Ihle JN, Yancopoulos GD. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science, 263, 92–95 (1994).
- 24) Stahl N, Farruggella TJ, Boulton TG, Zhong Z, Darnell JE Jr, Yancopoulos GD. Choice of STATs and other substrates specified by modular tyrosine-based motifs in cytokine receptors. Science, 267, 1349–1353 (1995).
- 25) Su F, Ren F, Rong Y, Wang Y, Geng Y, Wang Y, Feng M, Ju Y, Li Y, Zhao ZJ, Meng K, Chang Z. Protein tyrosine phosphatase Meg2 dephosphorylates signal transducer and activator of transcription 3 and suppresses tumor growth in breast cancer. Breast Cancer Res., 14, R38 (2012).
- 26) Huang FJ, Steeg PS, Price JE, Chiu WT, Chou PC, Xie K, Sawaya R, Huang S. Molecular basis for the critical role of suppressor of cytokine signaling-1 in melanoma brain metastasis. Cancer Res., 68, 9634–9642 (2008).
- 27) Brantley EC, Nabors LB, Gillespie GY, Choi YH, Palmer CA, Harrison K, Roarty K, Benveniste EN. Loss of protein inhibitors of activated STAT-3 expression in glioblastoma multiforme tumors: implications for STAT-3 activation and gene expression. Clin. Cancer Res., 14, 4694–4704 (2008).
- 28) Coe H, Jung J, Groenendyk J, Prins D, Michalak M. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem., 285, 6725–6738 (2010).
- 29) Meares GP, Liu Y, Rajbhandari R, Qin H, Nozell SE, Mobley JA, Corbett JA, Benveniste EN. PERK-dependent activation of JAK1 and STAT3 contributes to endoplasmic reticulum stress-induced inflammation. Mol. Cell. Biol., 34, 3911–3925 (2014).
- 30) Du XL, Yang H, Liu SG, Luo ML, Hao JJ, Zhang Y, Lin DC, Xu X, Cai Y, Zhan QM, Wang MR. Calreticulin promotes cell motility and enhances resistance to anoikis through STAT3-CTTN-Akt pathway in esophageal squamous cell carcinoma. Oncogene, 28, 3714–3722 (2009).
- 31) Matsuzaki Y, Besnard V, Clark JC, Xu Y, Wert SE, Ikegami M, Whitsett JA. STAT3 regulates ABCA3 expression and influences lamellar body formation in alveolar type II cells. Am. J. Respir. Cell Mol. Biol., 38, 551–558 (2008).
- 32) Bellido T, Borba VZ, Roberson P, Manolagas SC. Activation of the Janus kinase/STAT (signal transducer and activator of transcription) signal transduction pathway by interleukin-6-type cytokines promotes osteoblast differentiation. Endocrinology, 138, 3666–3676 (1997).
- 33) Uhlen M, Oksvold P, Fagerberg L, Lundberg E, Jonasson K, Forsberg M, Zwahlen M, Kampf C, Wester K, Hober S, Wernerus H, Björling L, Ponten F. Towards a knowledge-based Human Protein Atlas. Nat. Biotechnol., 28, 1248–1250 (2010).