Abstract
We previously reported that levels of long-chain fatty acids (FAs) including docosahexaenoic acids (DHA) increase in the hypothalamus of inflammatory pain model mice. However, the precise mechanisms underlying the increment of free fatty acids (FFAs) in the brain during inflammation remains unknown. In this study, we characterized FFAs released by inflammatory stimulation in rat primary cultured astrocytes, and tested the involvement of phospholipase A2 (PLA2) on these mechanisms. Lipopolysaccharide (LPS) stimulation significantly increased the levels of several FAs in the astrocytes. Under these conditions, mRNA expression of cytosolic PLA2 (cPLA2) and calcium-independent PLA2 (iPLA2) in LPS-treated group increased compared with the control group. Furthermore, in the culture media, the levels of DHA and arachidonic acid (ARA) significantly increased by LPS stimuli compared with those of a vehicle-treated control group whereas the levels of saturated FAs (SFAs), namely palmitic acid (PAM) and stearic acid (STA), did not change. In summary, our findings suggest that astrocytes specifically release DHA and ARA by inflammatory conditions. Therefore astrocytes might function as a regulatory factor of DHA and ARA in the brain.
Lipid is one of the necessary nutrients to keep homeostasis. Fatty acids (FAs) are the smallest unit that makes up Lipids. FAs are currently generating considerable attention as a major source of energy in the brain. In particular, a continuous supply of FAs is required for cellular metabolism, differentiation, and development of neuron.1–4) FAs served not only as cellular nutrients and cell membrane components but also as lipid-soluble signal molecules, suggesting that FAs play vital roles in a wide variety of physiological function.5,6) The central nervous system (CNS) is enriched in lipids7–10) and, in particular, brain consist of 20% polyunsaturated FAs (PUFAs) such as docosahexaenoic acid (DHA) and arachidonic acid (ARA). Furthermore, these PUFAs mainly relate the physiological function of the brain.
Astrocytes, one of the glial cells, assume a crucial role to maintain brain and neuronal function via releasing glutamate or neurotrophic factors. In CNS, astrocytes but not neurons are the main source of DHA and ARA. DHA and ARA are released from astroglial membranes under basal and stimulated conditions such as neurotransmitters, bladykinin, glutamate, ATP and thrombin and are supplied to the neurons.11–14) In our previous study, we demonstrated that hypothalamic long chain fatty acid (FA) levels increase in the early phase of complete Freund’s adjuvant (CFA)-induced inflammatory pain.15) The DHA level, in particular, is significantly increased compared with the control group. These increases correlated with an increase in glial fibrillary acidic protein (GFAP) expression in the hypothalamus of CFA-treated mice and were inhibited by the suppression of astrocyte proliferation induced by flavopiridol, a cyclin-dependent kinase inhibitor.16) However, it is unclear whether astrocytes really released DHA or other free fatty acids (FFAs) during inflammation.
In this study, we characterized FFAs that released by inflammatory stimulation in a rat primary cultured astrocytes using liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS), and tested the relationship phospholipase A2 (PLA2) and FFAs release on these mechanisms.
MATERIALS AND METHODS
Ethical Approval of the Study ProtocolThe present study was conducted in accordance with the Guiding Principles for the Care and Use of Laboratory Animals adopted by the Japanese Pharmacological Society. All experiments were approved by the ethics committee for animal experimentation of Kobe Gakuin University (approval number A15-11; Kobe, Japan).
AnimalsFemale Wistar rats (pregnant by 16 or 17 d) were obtained from Japan SLC, Inc. (Hamamatsu, Japan). Rats were housed in cages at 23–24°C with a 12-h light–dark cycle (lights phase, 08 : 00–20:00). Food and water were available ad libitum.
Primary Astrocyte PreparationAstrocytes were prepared from the cerebra of 0- to 2-d-old Wistar rats, as previously described.17) The isolated cells were seeded at 1×104 cells/cm2 in 75 cm2 culture flasks (Nunc, Roskilde, Denmark) and grown in minimal essential medium (Nissui Pharmaceutical Co., Ltd., Tokyo, Japan) supplemented with 10% fetal bovine serum (FBS) (Biosera, Boussens, France). To remove small process-bearing cells (mainly oligodendrocyte progenitors and microglia from the protoplasmic cell layer), the culture flasks were shaken at 250 rpm, 37°C overnight for 10–14 d after seeding. The monolayer cells were trypsinized (GIBCO BRL, Palo Alto, CA, U.S.A.) and seeded in 6-well culture plates (Nunc, Roskilde, Denmark) or on 15-mm glass coverslips (Matsunami Glass Ind., Ltd., Osaka, Japan) in 24-well culture plates (Nunc). At this stage, approximately 95% of the cells showed immunoreactivity for GFAP.
Cell Viability TestEffects of lipopolysaccharide (LPS) from Escherichia coli (Simga-Aldrich Co., St. Louis, MO, U.S.A.) on primary astrocytes were assessed with Cell Count Reagent SF (nacalai tesque, Kyoto, Japan). Primary astrocytes seeded at 1×105 cells/mL into 96-well plates. After incubated 24 h, primary astrocytes cultured with serum-free media for 48 h. Next, LPS was added into each well at various final concentrations and incubated for 24 h. After that, we exchanged with fresh culture media and reacted Cell Count Reagent SF for 1 h. Absorbance was measured at 450 nm and 630 nm using Corona grating microplate reader (SH-1200; Corona Electric Co., Ltd., Ibaraki, Japan).
Treatment of Primary Astrocytes in CultureBefore treatment, astrocytes in 6-well culture plates were incubated in serum-free medium for 48 h. LPS was minimally diluted using serum-free medium before treatment. After incubation in serum-free medium, the cells were treated with 100 or 1000 ng/mL LPS for 24 h.
Sample Preparation for the Determination of FFAs in Primary Cultured Astrocytes and Culture MediumThe FFAs in culture medium were freeze dried using FreeZone Freeze Dry system (LABCONCO FZ-12; Labconco, Kansas, MO, U.S.A.) and dissolve in methanol (Wako Pure Chemical Industries, Ltd., Osaka, Japan). The FFAs in primary astrocytes were extracted in methanol. Extracted solutions were centrifuged at 200×g for 5 min at 4°C and collected supernatant.
Comparative FFAs AnalysesThe sample were centrifuged at 15000×g for 15 min. The supernatant was collected and filtrated in sample vial for LC-MS/MS analysis. HPLC separation was performed with an Agilent 1290 Infinity LC (Agilent Technologies, California, U.S.A.) having a CAPCELL PAK UG120 column: 1.0 mm i.d.×150 mm (Shiseido, Tokyo, Japan). The mobile phases were A, 10 mM ammonium formate (pH 3.5) and B, acetonitrile. The eluting gradient was as follows: the column was equilibrated with 13% A, 13% A for 5 min, 13% A to 5% A in 5 min, 5% A for 5 min, 5% A to 13% A in 5 min, 13% A for 5 min. The flow rate was 0.2 mL/min. We used a QTRAP 4500 (AB SCIEX, Massachusetts, U.S.A.) to carry out quantitation. We operated the mass spectrometer in the negative-ion mode. The FFAs were quantified by selective multireaction monitoring (MRM) with a negative ionization mode. The ion spray voltage was −4500 V, the source temperature was 300°C, the declustering potential ranged −70 from −105 V and collision energy ranged −10 from −22 eV for the fragment ions. The peak of each FFAs was monitored by the product ion obtained from [M−H]-ion (i.e., m/z 255→255 for palmitic acid (PAM), m/z 279→279 for linoleic acid; LNA, m/z 281→281 for oleic acid; OLA, m/z 283→283 for stearic acid; STA, m/z 303→303 for ARA, m/z 327→327 for DHA). Each FFAs concentration was evaluated from each calibration curves by absolute calibration curve method.
Immunofluorescence StudiesPrimary astrocytes cultured on glass coverslips were washed with phosphate-buffered saline (PBS) containing 0.1% Triton X-100 (PBST) three times at 5 min intervals and incubated with blocking buffer (3% BSA in PBST) for 1 h at room temperature. The sections were then incubated with specific antibodies against GFAP (mouse monoclonal anti-GFAP, 1 : 10000; Millipore Corp., Billerica, MA, U.S.A.) overnight at 4°C at which time the antibody was diluted with reaction buffer (1% bovine serum albumin (BSA) in PBS). The next day, sections were washed with PBST and incubated at room temperature for 2 h with secondary antibody conjugated with AlexaFluor 594 (goat polyclonal anti-mouse immunoglobulin G (IgG) 1 : 200, Life Technologies, Inc., Carlsbad, CA, U.S.A.) at which time the secondary antibody was diluted with reaction buffer. After these sections were washed with PBST, we stained nuclear with 4′,6-diamidino-2-phenylindole (DAPI) (Boster Biological Tec., CA, U.S.A.) at room temperature for 10 min. Finally, sections washed with PBST and coverslipped with PermaFluor (Thermo Shandon Inc., Pittsburgh, PA, U.S.A.), and immunoreactivity was detected with a confocal fluorescence microscope (FV1000, Olympus Corporation, Tokyo, Japan). In the immunohistochemical control studies, no staining was detected when the corresponding primary or secondary antibody was omitted.
RNA Preparation and Real-Time PCRAstrocytes in 6-well culture plates were incubated in serum-free medium for 48 h before treatment with LPS. Total RNA was isolated from cultured astrocytes using the RNeasy mini kit (Qiagen, Hilden, Germany) and measured it by NanoDrop Lite Spectrophotometer (Thermo Fisher Scientific Inc., Massachusetts, U.S.A.). First-strand cDNA was synthesized from total RNA (500 ng) using the Primescript RT reagent kit (TaKaRa, Shiga, Japan). The mRNA levels were determined using real time-PCR with SYBR Green fluorescent probes using Light Cycler 96 (Roche, Basel, Switzerland). Expression of β-actin was used as an internal standard, and 20 µM each of primer and 20 ng of cDNA were used. The following primers were used: cPLA2 forward primer 5′-(AAC AGA GCA ACG AGA TGG)-3′ and reverse primer 5′-(AAC AGA GCA ACG AGA TGG)-3′18); iPLA2 forward primer 5′-(ATT GAT AAC AGA ACT CGA GC)-3′ and reverse primer 5′-(GAT GAA TCG GCT TCT GAG TA)-3′19); β-actin forward primer 5′-(GAA CCC TAA GGC CAA CCG TG)-3′ and reverse primer 5′-(TGG CAT AGA GGT CTT TAC GG)-3′,18) To determine each relative gene expression, the ΔΔCt method was employed and analyzed. To normalize target gene, β-actin used as a reference gene.
Statistical AnalysesData are expressed as the mean±standard error of the mean (S.E.M.). The statistical significance of differences were analyzed one-way ANOVA followed by Dunnett’s multiple comparison test. Differences resulting in p values <0.05 were considered statistically significant.
RESULTS
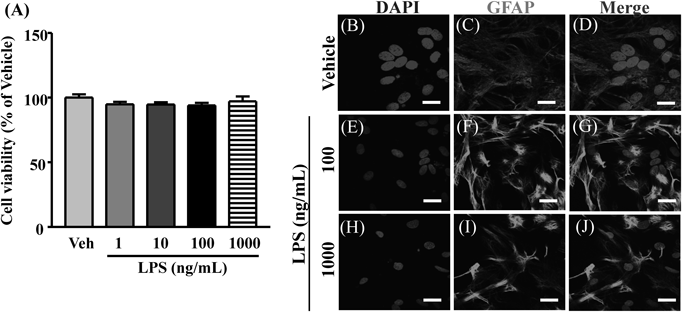
Structural Changes of Astrocytes Is Induced by LPS StimulationThe concentration level of LPS (1–1000 ng/mL) did not affect the cell viability (Fig. 1A). GFAP immunoreactivity increased 24 h after exposure to LPS (Figs. 1B–D). The morphology of the astrocytes also showed hypertrophy of the cell-body upon LPS stimulation compared with the vehicle-treated group (Figs. 1E–G). In contrast, astrocytes treated with vehicle did not show morphological changes.
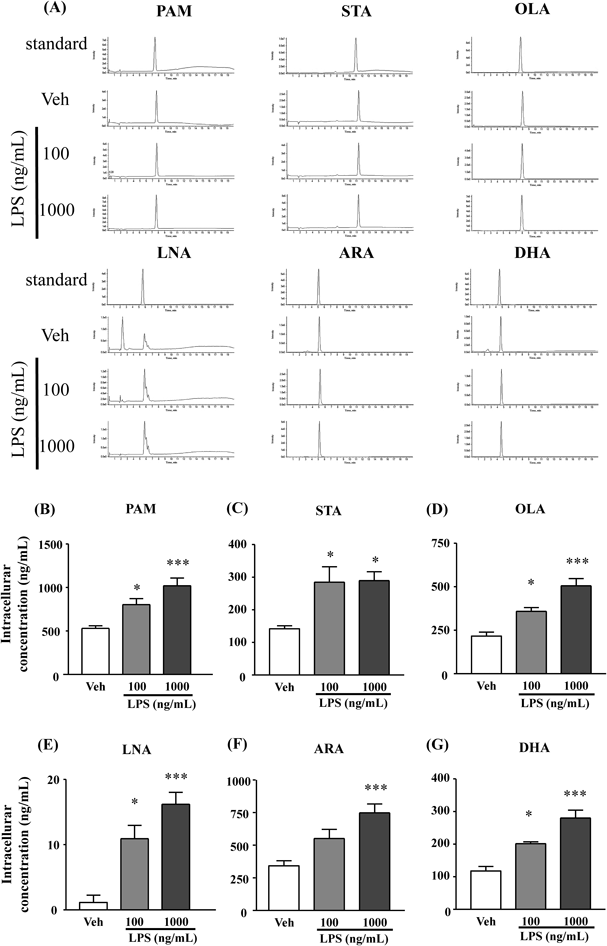
FFAs Levels Increased in Astrocytes after LPS Stimulation for 24 hTwenty-four hours after LPS (100 or 1000 ng/mL) stimulation, the levels of PAM, STA, OLA, and LNA dose-dependently increased compared with the vehicle-treated group (Figs. 2B–E). Furthermore, intracellular DHA and ARA levels significantly increased in a dose-dependent manner (Figs. 2F, G).
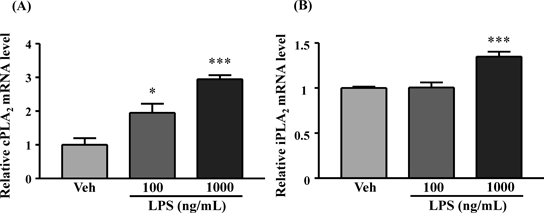
The cPLA2 and iPLA2 mRNA in Astrocyte Increased by LPS StimuliAfter exposure of astrocytes to LPS (100 and 1000 ng/mL) for 24 h, the expression of cPLA2 mRNA significantly and dose-dependently increased compared to the vehicle-treated group (Fig. 3A). Furthermore, iPLA2 mRNA significantly increased after stimulation with 1000 ng/mL LPS but not after stimulation with 100 ng/mL LPS compared with the vehicle-treated group (Fig. 3B).
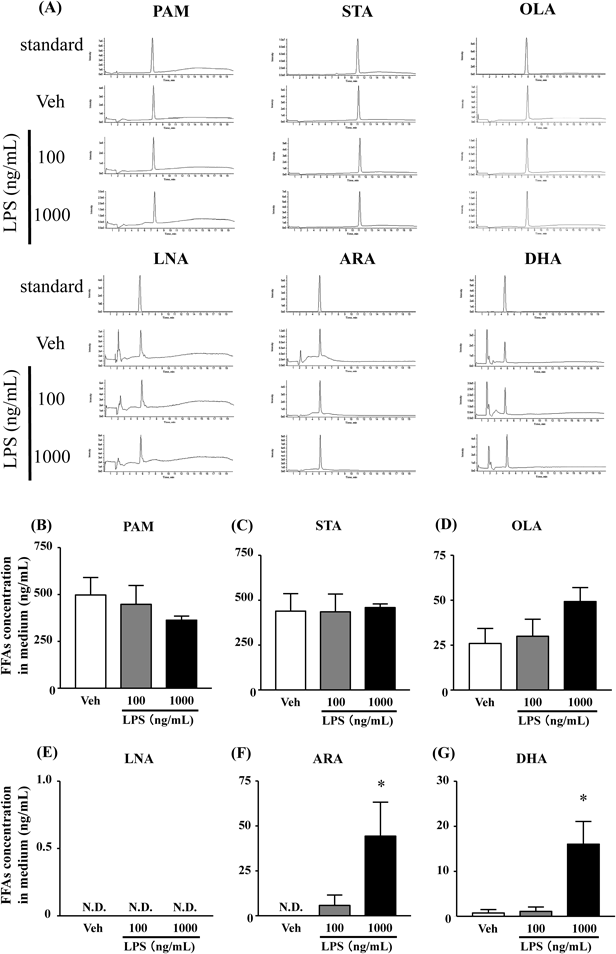
DHA and ARA Were Released by LPS Stimuli for 24 h in Culture MediaDHA, ARA, PAM, STA and OLA were detected in the culture media 24 h after stimulation with 100 or 1000 ng/mL LPS. Levels of the SFAs, PAM and STA as well as levels of the monounsaturated FA OLA did not change upon LPS stimulation (Figs. 4B–E). In contrast, levels of DHA and ARA considerably increased after LPS (1000 ng/mL) stimulation, compared with the vehicle control (Figs. 4F, G).
DISCUSSION
PUFAs are abundant in the phospholipid bilayer of brain and that they play important roles in brain function and maintenance of its structure.20–22) As previously reported, essential n-3 PUFAs, especially DHA, have beneficial actions in many inflammatory diseases.23–26) For example, in inflamed tissues at peripheral sites, DHA is released from immune cells such as neutrophils during the acute phase of the inflammatory process.27) Most important thing in this study is that LPS stimulation specifically facilitated the release of DHA and ARA from astrocytes although SFAs did not change.
However, in this study, the concentration of DHA and ARA released from astrocyte is nM order, and it is much lower than that of intracellular concentration. Another researchers suggest that DHA shows the beneficial effects in the level of µM in vivo.27) These differences might be due to increment of oxidative metabolites of DHA and ARA in culture. Because, it is well known that PUFAs are readily oxidized in the presence of oxygen. Another possibility is that n-3 FAs metabolites such as eicosapentaenoic acid (EPA)-derived (E-series) or DHA-derived (D-series) resolvins,28,29) which is known as a lipoxygenase and cytochrome P450 metabolites increased for 24 h treatment, whereas the level of DHA is decreased. And also, ARA is easily metabolite via cyclooxygenase or lipoxygenase, and result in induction of the production of pro-inflammatory factors such as prostaglandin E2 and leukotriene,30) and ARA-derived eicosanoids lipoxin which known modulate inflammation to resolve the pro-inflammatory process.29,31) However, it is unclear whether the concentration of ARA and DHA released from astrocytes has beneficial effect such as an anti-inflammation effect or not. Therefore, our results indicate that astrocytes might be involved in control of brain FFAs composition respond to LPS stimulation. Based on our previous reports,32,33) it is thought that these FFAs released from astrocytes might maintain a neuronal function as a ligand of GPR40/FFAR1, one of a FFAs receptors.34)
Next, we examined how FFAs, especially DHA and ARA, increase in astrocytes after LPS stimulation. It is widely accepted that the main site of storage for DHA and ARA is the sn-2 position of membrane phospholipids35) and that enzymes responsible for their release are intracellular PLA2 family.36,37) Several groups of PLA2 enzymes have been identified in the mammalian brain,38–41) and their specificity have been characterized in previous studies. ARA is selectively hydrolyzed by cPLA2 in a calcium-dependent manner, whereas DHA in sn-2 position is selectively hydrolyzed in a calcium-independent manner by iPLA2.42) iPLA2 and cPLA2 have been reported to be expressed in different brain regions, including astrocytes.43–45) After LPS stimulation, cPLA2 and iPLA2 mRNA level were concomitantly increased at the timing which DHA or ARA increase in the media. These results suggesting that the increasing of DHA and ARA in the media with LPS might be caused increment of intracellular FFA through increasing of intracellular cPLA2 and iPLA2.
In conclusion, we demonstrated that astrocytes release DHA and ARA during the inflammatory state to control inflammation processes in the CNS. Therefore, our findings suggest that astrocytes-derived PUFAs contribute to the maintenance of brain function and its environment and perform as one of the modulators of neuronal function under inflammatory conditions.
Acknowledgments
This work was supported by the Takeda Science Foundation and a Grant-in-Aid for Scientific Research (C) (15K10566) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Katakura M, Hashimoto M, Okui T, Shahdat HM, Matsuzaki K, Shido O. Omega-3 polyunsaturated fatty acids enhance neuronal differentiation in cultured rat neural stem cells. Stem Cells Int., 2013, 490476 (2013).
- 2) Wójcik C, Lohe K, Kuang C, Xiao Y, Jouni Z, Poels E. Modulation of adipocyte differentiation by omega-3 polyunsaturated fatty acids involves the ubiquitin-proteasome system. J. Cell. Mol. Med., 18, 590–599 (2014).
- 3) Ma D, Zhang M, Larsen CP, Xu F, Hua W, Yamashima T, Mao Y, Zhou L. DHA promotes the neuronal differentiation of rat neural stem cells transfected with GPR40 gene. Brain Res., 1330, 1–8 (2010).
- 4) Maximin E, Langelier B, Aïoun J, Al-Gubory KH, Bordat C, Lavialle M, Heberden C. Fatty acid binding protein 7 and n-3 poly unsaturated fatty acid supply in early rat brain development. Dev. Neurobiol., 76, 287–297 (2016).
- 5) Tessaro FHG, Ayala TS, Martins JO. Lipid mediators are critical in resolving inflammation: a review of the emerging roles of eicosanoids in diabetes mellitus. Biomed. Res. Int., 2015, 568408 (2015).
- 6) Hirabayashi T, Murayama T, Shimizu T. Regulatory mechanism and physiological role of cytosolic phospholipase A2. Biol. Pharm. Bull., 27, 1168–1173 (2004).
- 7) Kim H-Y, Huang BX, Spector AA. Phosphatidylserine in the brain: metabolism and function. Prog. Lipid Res., 56, 1–18 (2014).
- 8) O’Brien JS, Fillerup DL, Mead JF. Quantification and fatty acid and fatty aldehyde composition of ethanolamine, choline, and serine glycerophosphatides in human cerebral grey and white matter. J. Lipid Res., 5, 329–338 (1964).
- 9) Breckenridge WC, Gombos G, Morgan IG. The lipid composition of adult rat brain synaptosomal plasma membranes. Biochim. Biophys. Acta, 266, 695–707 (1972).
- 10) Cotman C, Blank ML, Moehl A, Snyder F. Lipid composition of synaptic plasma membranes isolated from rat brain by zonal centrifugation. Biochemistry, 8, 4606–4612 (1969).
- 11) Stella N, Tence M, Glowinski J, Premont J. Glutamate-evoked release of arachidonic acid from mouse brain astrocytes. J. Neurosci., 14, 568–575 (1994).
- 12) Stella N, Estellés A, Siciliano J, Tencé M, Desagher S, Piomelli D, Glowinski J, Prémont J. Interleukin-1 enhances the ATP-evoked release of arachidonic acid from mouse astrocytes. J. Neurosci., 17, 2939–2946 (1997).
- 13) Moore SA. Polyunsaturated fatty acid synthesis and release by brain-derived cells in vitro. J. Mol. Neurosci., 16, 195–200, discussion, 215–221 (2001).
- 14) Strokin M, Sergeeva M, Reiser G. Docosahexaenoic acid and arachidonic acid release in rat brain astrocytes is mediated by two separate isoforms of phospholipase A2 and is differently regulated by cyclic AMP and Ca2+. Br. J. Pharmacol., 139, 1014–1022 (2003).
- 15) Nakamoto K, Nishinaka T, Sato N, Mankura M, Koyama Y, Kasuya F, Tokuyama S. Hypothalamic GPR40 signaling activated by free long chain fatty acids suppresses CFA-induced inflammatory chronic pain. PLoS ONE, 8, e81563 (2013).
- 16) Di Giovanni S, Movsesyan V, Ahmed F, Cernak I, Schinelli S, Stoica B, Faden AI. Cell cycle inhibition provides neuroprotection and reduces glial proliferation and scar formation after traumatic brain injury. Proc. Natl. Acad. Sci. U.S.A., 102, 8333–8338 (2005).
- 17) Koyama Y, Maebara Y, Hayashi M, Nagae R, Tokuyama S, Michinaga S. Endothelins reciprocally regulate VEGF-A and angiopoietin-1 production in cultured rat astrocytes: implications on astrocytic proliferation. Glia, 60, 1954–1963 (2012).
- 18) Lin CC, Hsieh HL, Liu SW, Tseng HC, Hsiao LD, Yang CM. BK induces cPLA2 expression via an autocrine loop involving COX-2-derived PGE2 in rat brain astrocytes. Mol. Neurobiol., 51, 1103–1115 (2015).
- 19) Strokin M, Sergeeva M, Reiser G. Prostaglandin synthesis in rat brain astrocytes is under the control of the n-3 docosahexaenoic acid, released by group VIB calcium-independent phospholipase A2. J. Neurochem., 102, 1771–1782 (2007).
- 20) Contreras MA, Greiner RS, Chang MC, Myers CS, Salem N Jr, Rapoport SI. Nutritional deprivation of alpha-linolenic acid decreases but does not abolish turnover and availability of unacylated docosahexaenoic acid and docosahexaenoyl-CoA in rat brain. J. Neurochem., 75, 2392–2400 (2000).
- 21) Rapoport SI. Arachidonic acid and the brain. J. Nutr., 138, 2515–2520 (2008).
- 22) Steenbergen R, Nanowski TS, Nelson R, Young SG, Vance JE. Phospholipid homeostasis in phosphatidylserine synthase-2-deficient mice. Biochim. Biophys. Acta, 1761, 313–323 (2006).
- 23) Yan Y, Jiang W, Spinetti T, Tardivel A, Castillo R, Bourquin C, Guarda G, Tian Z, Tschopp J, Zhou R. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity, 38, 1154–1163 (2013).
- 24) Figueroa JD, Cordero K, Baldeosingh K, Torrado AI, Walker RL, Miranda JD, De Leon M. Docosahexaenoic acid pretreatment confers protection and functional improvements after acute spinal cord injury in adult rats. J. Neurotrauma, 29, 551–566 (2012).
- 25) McNamara RK, Jandacek R, Rider T, Tso P, Cole-Strauss A, Lipton JW. Omega-3 fatty acid deficiency increases constitutive pro-inflammatory cytokine production in rats: relationship with central serotonin turnover. Prostaglandins Leukot. Essent. Fatty Acids, 83, 185–191 (2010).
- 26) Marion-Letellier R, Savoye G, Ghosh S. Polyunsaturated fatty acids and inflammation. IUBMB Life, 67, 659–667 (2015).
- 27) Bravo E, Napolitano M, Valentini SB, Quattrucci S. Neutrophil unsaturated fatty acid release by GM-CSF is impaired in cystic fibrosis. Lipids Health Dis., 9, 129 (2010).
- 28) Schwab JM, Chiang N, Arita M, Serhan CN. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature, 447, 869–874 (2007).
- 29) Spite M, Serhan CN. Novel lipid mediators promote resolution of acute inflammation: impact of aspirin and statins. Circ. Res., 107, 1170–1184 (2010).
- 30) Yagami T. Cerebral arachidonate cascade in dementia: Alzheimer’s disease and vascular dementia. Curr. Neuropharmacol., 4, 87–100 (2006).
- 31) Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol., 8, 349–361 (2008).
- 32) Nakamoto K, Nishinaka T, Sato N, Aizawa F, Yamashita T, Mankura M, Koyama Y, Kasuya F, Tokuyama S. The activation of supraspinal GPR40/FFA1 receptor signalling regulates the descending pain control system. Br. J. Pharmacol., 172, 1250–1262 (2015).
- 33) Nakamoto K, Nishinaka T, Ambo A, Mankura M, Kasuya F, Tokuyama S. Possible involvement of β-endorphin in docosahexaenoic acid-induced antinociception. Eur. J. Pharmacol., 666, 100–104 (2011).
- 34) Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR Jr, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem., 278, 11303–11311 (2003).
- 35) Yabuuchi H, O’Brien JS. Positional distribution of fatty acids in glycerophosphatides of bovine gray matter. J. Lipid Res., 9, 65–67 (1968).
- 36) Smani T, Zakharov SI, Leno E, Csutora P, Trepakova ES, Bolotina VM. Ca2+-independent phospholipase A2 is a novel determinant of store-operated Ca2+ entry. J. Biol. Chem., 278, 11909–11915 (2003).
- 37) Sun GY, Chuang DY, Zong Y, Jiang J, Lee JC, Gu Z, Simonyi A. Role of cytosolic phospholipase A2 in oxidative and inflammatory signaling pathways in different cell types in the central nervous system. Mol. Neurobiol., 50, 6–14 (2014).
- 38) Farooqui AA, Horrocks LA. Brain phospholipases A2: a perspective on the history. Prostaglandins Leukot. Essent. Fatty Acids, 71, 161–169 (2004).
- 39) Chiba H, Michibata H, Wakimoto K, Seishima M, Kawasaki S, Okubo K, Mitsui H, Torii H, Imai Y. Cloning of a gene for a novel epithelium-specific cytosolic phospholipase A2, cPLA2delta, induced in psoriatic skin. J. Biol. Chem., 279, 12890–12897 (2004).
- 40) Underwood KW, Song C, Kriz RW, Chang XJ, Knopf JL, Lin LL. A novel calcium-independent phospholipase A2, cPLA2-gamma, that is prenylated and contains homology to cPLA2. J. Biol. Chem., 273, 21926–21932 (1998).
- 41) Yang HC, Mosior M, Ni B, Dennis EA. Regional distribution, ontogeny, purification, and characterization of the Ca2+-independent phospholipase A2 from rat brain. J. Neurochem., 73, 1278–1287 (1999).
- 42) Rosa AO, Rapoport SI. Intracellular- and extracellular-derived Ca2+ influence phospholipase A(2)-mediated fatty acid release from brain phospholipids. Biochim. Biophys. Acta, 1791, 697–705 (2009).
- 43) Xu J, Yu S, Sun AY, Sun GY. Oxidant-mediated AA release from astrocytes involves cPLA(2) and iPLA(2). Free Radic. Biol. Med., 34, 1531–1543 (2003).
- 44) Ong WY, Sandhya TL, Horrocks LA, Farooqui AA. Distribution of cytoplasmic phospholipase A2 in the normal rat brain. J. Hirnforsch., 39, 391–400 (1999).
- 45) Ong WY, Yeo JF, Ling SF, Farooqui AA. Distribution of calcium-independent phospholipase A2 (iPLA 2) in monkey brain. J. Neurocytol., 34, 447–458 (2005).