Abstract
Ursodeoxycholic acid (UDCA) is a hydrophilic bile acid that possesses many pharmacological effects, including increasing bile flow, changing the hydrophobicity of the bile acid pool, and modulation of the immune response. UDCA has been approved for treating cholestatic liver disease, such as primary biliary cholangitis. However, several unanticipated severe side effects of UDCA are observed in cholestatic patients, and its pharmacological benefits remain controversial. We reported that ezrin-knockdown (Vil2kd/kd) mice exhibited severe hepatic injury because of a functional disorder in bile duct fluidity and alkalinity regulation, resembling human intrahepatic cholestatic disease. Here we used Vil2kd/kd mice as a cholestatic model to investigate the pharmacological effects of UDCA. We investigated the effects of oral and parenteral administration of UDCA on Vil2kd/kd mice. In Vil2kd/kd mice, fed a 0.5% (w/w) UDCA diet for 3 weeks, hepatic injury was exacerbated, although oral administration of a lower dose of UDCA slightly improved hepatic function in Vil2kd/kd mice. On the other hand, intraperitoneal administration of UDCA (50 mg/kg/d) ameliorated hepatic function and markedly reduced periductal fibrosis and cholangiocyte proliferation in Vil2kd/kd mice although biliary pH and HCO3− concentration were not improved. The expression levels of inflammatory and profibrotic genes were also significantly decreased in these mice. Furthermore, UDCA prevented cholangiocytes from hydrophobic bile acid-induced cytotoxicity independent of extracellular pH in in vitro experiments. These results suggest that an appropriate dosage of UDCA can ameliorate the intrahepatic cholestasis in Vil2kd/kd mice without changing the biliary bicarbonate secretion.
Ursodeoxycholic acid (UDCA) is a steroid bile acid widely utilized for treating cholestatic liver disease and dissolving gallstones. UDCA was first identified in bear bile, but it is also present as a minor constituent in human bile. It is one of the secondary bile acids that are formed by bacterial enzymes in the intestine. UDCA is currently the only drug approved for treating patients with primary biliary cirrhosis (PBC) although there is still controversy surrounding its therapeutic value in patients with primary sclerosing cholangitis (PSC).1) UDCA is generally given by oral administration and is absorbed from the ileum. The apical sodium-dependent bile acid transporter (ASBT), which is highly expressed in the apical membrane of the ileum, contributes to the enterohepatic circulation of bile acids, including UDCA.2) The absorbed bile acids are then transferred to the basolateral side by the cytoplasmic ileal bile acid binding protein (IBABP)3) and secreted into the blood stream as a heterodimer of organic solute transporter alpha (OSTα) and OSTβ.4) The abnormal function of these transporters in the intestine causes bile acid malabsorption, which is often found in various gastrointestinal diseases.2,5) Therefore, absorption and therapeutic effects of orally administered UDCA from intestine are influenced by the ileal conditions. Because approximately 75% of PSC patients have inflammatory bowel disease (IBD) and ulcerative colitis (UC),6) ileal absorption of UDCA might be impaired.
We previously reported that knockdown of ezrin, a cytoskeletal adaptor protein predominantly expressed in intrahepatic bile ducts of the liver,7) causes severe hepatic injury by impairing the bile duct function. In the bile ducts, ezrin regulates the apical membrane localizations and activity of the cystic fibrosis transmembrane conductance regulator (CFTR) via interaction with another scaffold protein, Na+/H+ exchanger regulatory factor 1 (NHERF1). Abnormal membrane localizations of anion exchanger-2 (AE2) and aquaporin-1 (AQP1) were also observed in ezrin-knockdown (Vil2kd/kd) mice.7) Functional coupling between CFTR and AE2 contributes to the formation of biliary bicarbonate umbrella, which plays an important role in protecting the biliary epithelium from cytotoxic bile acids.8) In Vil2kd/kd mice, biliary bicarbonate umbrella formation is impaired. To date, several animal models showing individual characteristics of PBC and PSC were reported.9) AE2 knockout (Ae2a,b−/−) mice, and CFTR knockout (cftr−/−) mice show cholestatic liver damage similar with PBC and PSC, respectively,10,11) possibly due to the dysfunction of biliary bicarbonate umbrella formation although the hepatic injury was mild and observed after 6 months of age. On the other hand, Vil2kd/kd mice show more severe phenotype in early life compared to both Ae2a,b−/− and cftr−/− mice, due to the mislocalization of these transporters in the bile ducts. Thus, Vil2kd/kd mice will be useful for investigating the therapeutic potential of UDCA and other substances on cholestatic disease attributed to the dysfunction of biliary bicarbonate umbrella formation. In this study, we investigated the efficacy of UDCA on the bicarbonate umbrella dysfunction using Vil2kd/kd mice.
MATERIALS AND METHODS
Animal ExperimentsVil2kd/kd mice were generated as described previously.12) Twelve-week-old sex-matched, wild type (WT) and Vil2kd/kd mice were used in this study. All work with animals was approved by the Animal Ethics Committee of Ritsumeikan University, Japan. Blood was obtained via heart puncture under appropriate anesthesia, and the plasma was separated by centrifugation at 1000×g for 10 min at 4°C. Concentrations of plasma aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were determined using DRI-CHEM 4000i (FUJIFILM, Tokyo, Japan).
Protocol 1WT and Vil2kd/kd mice were divided into two groups. Mice were fed a normal diet (CE-2: CLEA Japan Inc., Tokyo, Japan) or CE-2 containing 0.5% w/w UDCA (Wako Pure Chemical Industries, Ltd., Osaka, Japan), and drinking water ad libitum for 3 weeks. Every week, peripheral blood was collected and ALT and AST were measured. At the day of sacrifice, trunk blood was collected by heart puncture, and liver tissue was collected for histological and quantitative reverse transcription (RT)-PCR analysis.
Protocol 2UDCA dissolved in saline (50 mg/kg body weight (BW)/d, or 100 mg/kg BW/d), or saline as a vehicle was administrated by gavage for 3 weeks in mice fed a normal diet and drinking water ad libitum.
Protocol 3A UDCA solution (DS Pharma-Animal Health, Osaka, Japan) was diluted in saline. Saline as a vehicle or 25, 50, and 100 mg/kg BW/d of UDCA were administered intraperitoneally for 3 weeks.
Liver Histology and Immunofluorescence StainingHematoxylin and Eosin (H&E) or Sirius Red staining were performed as described previously.7) The mouse liver was fixed with 4% paraformaldehyde (PFA) in 0.1 M phosphate buffer (PB) (pH 7.4), embedded in paraffin, and then sectioned (4 µm). After deparaffinization and rehydration, the sections were used for H&E or Sirius Red staining, which was performed using a Picrosirius Red staining kit (Polysciences Inc., Warrington, PA, U.S.A.).
Real Time RT-PCRReal time RT-PCR was performed as described previously.7) The primers used for PCR amplification are listed as below. Procollagen 1: sense, 5′-GCA GGG TTC CAA CGA TGT TG-3′, antisense, 5′-GCA GCC ATC GAC TAG GAC AGA-3′. Transforming growth factor-β (TGF-β): sense, 5′-TCG ACA TGG AGC TGG TGA AA-3′, antisense, 5′-CTG GCG AGC CTT AGT TTG GA-3′. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH): sense, 5′-TGT GTC CGT CGT GGA TCT GA-3′, antisense, 5′-TTG CTG TTG AAG TCG CAG GAG-3′. Tumor necrosis factor (TNF)-α: sense, 5′-TAT GGC CCA GAC CCT CAC A-3′, antisense, 5′-GGA GTA GAC AAG GTA CAA CCC ATC-3′, interleukin (IL)-1β: sense, 5′-TCC AGG ATG AGG ACA TGA GCA C-3′, antisense, 5′-GAA CGT CAC ACA CCA GCA GGT TA-3′.
Bile Flow MeasurementBile flow was measured as described previously.7) Briefly, under anesthetized conditions, bile was collected after surgical ligation of the common bile duct and cannulation of the gallbladder using polyethylene tube (PE-10). Bile was collected for 30 min. Bile flow rate was assessed gravimetrically assuming a density of 1 g/mL.
Measurement of Biliary pH and Bicarbonate ConcentrationBiliary pH and HCO3− concentration were measured as previously reported.7) Briefly, approximately 100 µL of bile was collected by the cannulation of polyethylene tube in to the gallbladder. Immediately after collecting bile, biliary pH and bicarbonate concentration was measured by blood gas meter ABL555 (Radiometer).
Cell Culture and Cell Viability AssayNormal mouse cholangiocytes (NMCs) immortalized through the introduction of the Simian virus 40 large T antigen gene were used. NMCs were maintained in RITC 80-7 medium containing 10% fetal bovine serum (FBS), 1% penicillin and streptomycin, and 2 mM L-glutamine at 37°C in a humidified 5% CO2 incubator as previously reported.7) To induce cholangiotoxicity, confluent NMCs were cultured in the presence of chenodeoxycholic acid (CDCA), or dimethyl sulfoxide (DMSO) as control at indicated concentration and a pH of 8.0, 7.4, 7.0, and 6.5 for 2 h. Cell viability was measured by CellTiter blue assay (Promega), which is based on the ability of living cells to convert a redox dye (resazurin) into a fluorescent end product (resorufin). The fluorescent intensity was measured by fluorometer.
Statistical AnalysisStatistical analysis was performed by Student’s t-test or one way ANOVA with the Turkey’s post test in GraphPad Prism 6.0 (San Diego, CA, U.S.A.) with p<0.05, and p<0.01 being considered statistically significant.
RESULTS
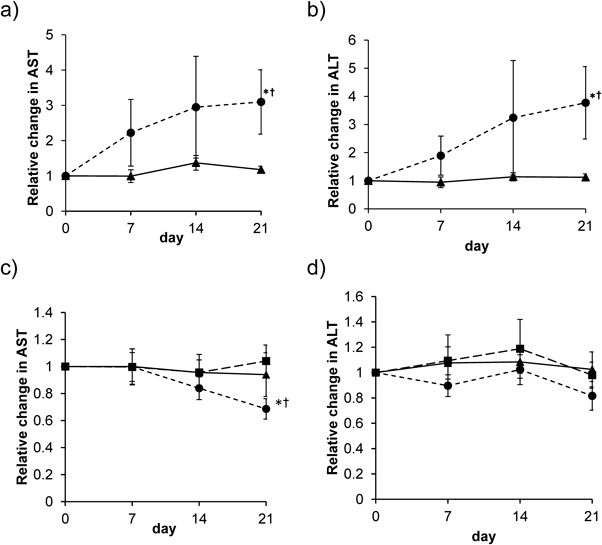
Oral Administration of UDCA to WT and Vil2kd/kd MiceTo examine the therapeutic effects of UDCA on Vil2kd/kd mice, we prepared a UDCA containing-diet in accordance with a previous report by Fickert et al.13) WT and Vil2kd/kd mice were fed a 0.5% (w/w) UDCA diet or a control diet for 3 weeks. In WT mice, we observed no changes based on the treatment with the 0.5% (w/w) UDCA diet during the experimental period (Table 1). However, Vil2kd/kd mice fed a 0.5% (w/w) UDCA diet exhibited a marked deterioration in hepatic function, similar to as has been previously reported in Mdr2-knockout mice.14) Plasma concentrations of AST and ALT gradually increased during the 3 weeks in Vil2kd/kd mice treated with the 0.5% (w/w) UDCA diet (Table 1), at which point they were 3-fold higher than before administration (Figs. 1a, b). Feeding of a 0.5% (w/w) UDCA diet is equivalent to a dosage of 500 mg to 1 g/kg BW/d UDCA of oral administration in mice. In humans, UDCA is generally used at range from 10 to 15 mg/kg/BW/d.1) Because a 0.5% (w/w) UDCA diet is considered a high dosage for mice, we gave an oral administration at a lower dosage of UDCA at 50 mg/kg BW/d and 100 mg/kg/d by gavage. The therapeutic effect was weak, but AST decreased by about 20% in Vil2kd/kd mice treated with 50 mg/kg BW/d UDCA (Table 2, and Fig. 1c), although ALT did not significantly change (Table 2, and Fig. 1d). On the other hand, administration of 100 mg/kg BW/d UDCA shows no obvious therapeutic efficacy. These data suggest that dosage control of UDCA is important for treating intrahepatic cholestasis in Vil2kd/kd mice, and a higher dosage of UDCA has a toxic effect on cholestatic mice14) although it has no adverse effect in WT mice.
Table 1. Parameters of Liver Function in Mice Administered with a UDCA Diet
| WT | 0 | 7 | 14 | 21 (d) |
|---|
| Control diet |
| AST, U/L | 57.0±5.7 | 74.0±11.6 | 57.6±5.2 | 68.6±10.3 |
| ALT, U/L | 25.3±2.0 | 37.0±4.8 | 9.4±8.0 | 36.6±3.9 |
| 0.5% (w/w) UDCA diet |
| AST, U/L | 53.7±4.2 | 60.0±6.4 | 51.5±3.7 | 54.0±3.8 |
| ALT, U/L | 30.5±5.6 | 40.2±4.7 | 42.8±5.6 | 36.7±6.9 |
| Vil2kd/kd |
|---|
| Control diet |
| AST, U/L | 141.0±8.5 | 194.7±56.2 | 189.6±20.2 | 176.7±15.0 |
| ALT, U/L | 137.7±11.6 | 130.6±27.4 | 148.2±18.7 | 162.2±24.9 |
| 0.5% (w/w) UDCA diet |
| AST, U/L | 134.3±20.7 | 252.3±79.8 | 282.0±133.9 | 358.0±77.2*† |
| ALT, U/L | 152.8±35.6 | 313.7±113.8 | 410.3±218.9 | 476.5±131.5*† |
* p<0.05, vs. day 0 in each group of mice, † p<0.05 vs. Vil2kd/kd mice fed control diet on the same day. n=6 in each group.
Table 2. Parameters of Liver Function in Mice Administered with Oral UDCA
| Vil2kd/kd | 0 | 7 | 14 | 21 (d) |
|---|
| Saline |
| AST, U/L | 151.9±21.9 | 135.5±12.4 | 133.4±12.9 | 146.4±7.8 |
| ALT, U/L | 136.0±23.4 | 129.9±14.7 | 137.6±16.9 | 120.1±7.4 |
| UDCA (50 mg/kg/d) |
| AST, U/L | 158.9±14.0 | 150.3±21.7 | 131.1±16.7 | 103.8±8.8*† |
| ALT, U/L | 154.3±21.1 | 119.4±12.1 | 131.9±8.4 | 111.3±7.4 |
| UDCA (100 mg/kg/d) |
| AST, U/L | 142.0±15.5 | 133.6±14.1 | 130.7±15.5 | 151.4±25.0 |
| ALT, U/L | 168.8±24.9 | 162.6±15.1 | 185.0±35.0 | 161.6±15.9 |
* p<0.05, vs. day 0 in each group of mice, † p<0.05 vs. Vil2kd/kd mice treated with saline on the same day. n=8 in each group.
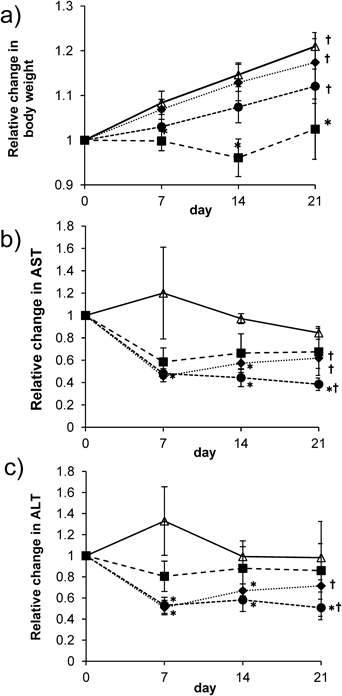
The therapeutic efficacy of oral UDCA was weaker than we had expected. Accordingly, we tried to alter the UDCA administration route as oral administration is often influenced by the condition of the gastrointestinal tract. As previously reported, Vil2kd/kd mice exhibited changes in protein expression levels in the gastrointestinal tract.12,15,16) We therefore examined the therapeutic efficacy of i.p. administration of UDCA by comparing dosages of 100 mg/kg BW/d (UDCA-100 i.p.), 50 mg/kg BW/d (UDCA-50 i.p.), and 25 mg/kg BW/d of UDCA (UDCA-25 i.p.) dissolved in saline, and using saline as a control, for 3 consecutive weeks. 100 mg/kg/d of UDCA administration showed no toxic effects on plasma AST, and ALT in WT mice (Supplementary Table 1). However, Vil2kd/kd mice receiving the UDCA-25 i.p. or UDCA-50 i.p. regimen had a significant increase in BW, whereas mice receiving the UDCA-100 i.p. regimen had no BW gain during the 3 weeks (Fig. 2a). Body weight gain of Vil2kd/kd mice, receiving the saline or UDCA-50 i.p. regimen were similar with those of WT during experimental periods (1.03±0.01 at day 21). In Vil2kd/kd mice on UDCA-100 i.p., plasma AST level was slightly reduced although the ALT level was not altered. (Table 3 and Figs. 2b, c). Some of the mice receiving UDCA-100 i.p. died within 3 weeks (data not shown) although WT mice receiving UDCA-100 i.p. did not show any abnormalities in the parameters of liver function (Supplementary Table 1). On the other hand, a lower dose of administration of UDCA provided a significant improvement in liver function in Vil2kd/kd mice. Administration of UDCA-50 i.p. was more effective at improving liver function compared with UDCA-25 i.p. in Vil2kd/kd mice (Fig. 2, Table 3).
Table 3. Parameters of Liver Function in Mice Administered with UDCA Intraperitoneally
| Vil2kd/kd | 0 | 7 | 14 | 21 (d) |
|---|
| Saline |
| AST, U/L | 206.4±26.4 | 211.8±61.6 | 185.3±13.4 | 160.0±6.4 |
| ALT, U/L | 171.4±22.5 | 195.5±45.7 | 180.3±8.6 | 176.0±2.0 |
| UDCA (25 mg/kg/d) |
| AST, U/L | 205.9±25.8 | 91.9±14.1* | 119.6±15.7* | 115.0±16.1* |
| ALT, U/L | 159.6±11.1 | 82.9±11.7* | 121.3±17.1 | 119.3±10.5*† |
| UDCA (50 mg/kg/d) |
| AST, U/L | 257.7±60.0 | 113.4±16.5 | 113.1±24.6 | 97.7±9.6 |
| ALT, U/L | 198.6±26.2 | 97.0±10.6* | 133.4±34.3 | 101.6±10.8*† |
| UDCA (100 mg/kg/d) |
| AST, U/L | 231.5±49.6 | 123.2±22.1 | 108.0±17.2* | 101.0±20.9 |
| ALT, U/L | 161.3±16.2 | 141.4±22.5 | 137.0±27.7 | 129.0±52.3 |
* p<0.05, vs. saline treated Vil2kd/kd mice on each day. †p<0.05, vs. day 0 in each mice. n=5–7.
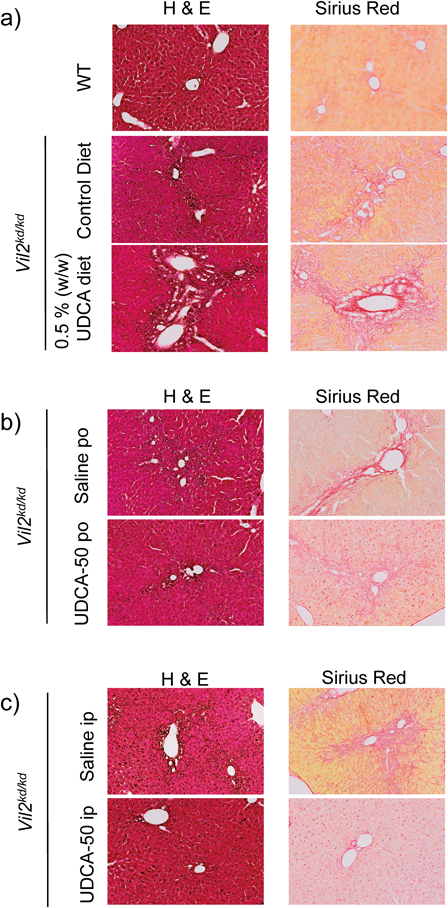
To observe the histological alterations after UDCA treatment in Vil2kd/kd mice, H&E staining and Sirius red staining were performed. H&E staining demonstrated exacerbated hepatic injury in Vil2kd/kd mice treated with the 0.5% (w/w) UDCA diet (Fig. 3a). Sirius red staining indicated that periductal fibrosis and abnormal cholangiocyte proliferation had progressed in Vil2kd/kd mice treated with the 0.5% (w/w) UDCA diet (Fig. 3a). Oral administration of UDCA at a lower dose showed signs of slight recovery in periductal fibrosis and cholangiocyte proliferation (Fig. 3b). These results correspond to the changes in plasma AST and ALT concentrations in Vil2kd/kd mice (Fig. 1). On the other hand, administration of UDCA-50 i.p. markedly improved periductal fibrosis and cholangiocyte proliferation (Fig. 3c), suggesting that an i.p. injection of 50 mg/kg BW/d UDCA is appropriate for treating intrahepatic cholestasis in Vil2kd/kd mice.
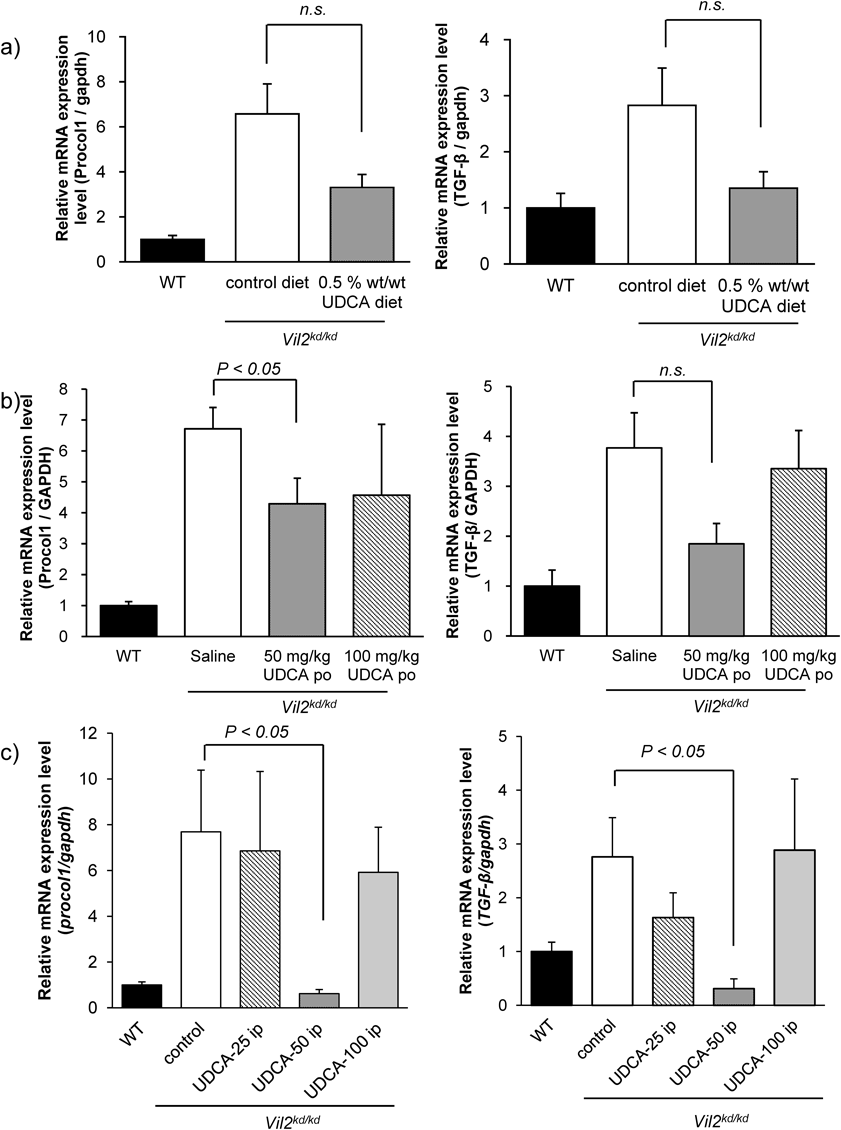
The Effects of Administration of UDCA on the Inflammatory- and Fibrosis-Related Gene Expression in Vil2kd/kd MiceTo investigate how UDCA improves intrahepatic cholestasis in Vil2kd/kd mice, we performed quantitative RT-PCR analyses and assessed the inflammatory cytokines and profibrotic factor expression levels. In Vil2kd/kd mice fed a 0.5% (w/w) UDCA diet, mRNA expression levels of TGF-β and procollagen 1 decreased, but were not significantly altered compared with the control mice (Fig. 4a). Oral administration of UDCA at 50 mg/kg, but not at 100 mg/kg, also decreased mRNA levels of TGF-β and procollagen 1. The expression level of procollagen 1 mRNA was significantly decreased compared with that in saline-treated Vil2kd/kd mice, although the expression level was still higher than that in WT mice (Fig. 4b). On the other hand, procollagen 1 expression was markedly decreased in UDCA-50 i.p.-treated Vil2kd/kd mice, whereas UDCA-25 i.p.- or UDCA-100 i.p.-treated mice exhibited higher expression levels of procollagen 1 than the WT mice. TGF-β mRNA expression was also significantly decreased in UDCA-50 i.p.-treated Vil2kd/kd mice, and was less than that seen in WT mice. TGF-β expression in Vil2kd/kd mice with UDCA-100 i.p. was kept at a similar level with those of vehicle treated Vil2kd/kd mice (Fig. 4c). The expression levels of TNF-α, and IL-1β were also investigated. Significant reduction of TNF-α was observed in both UDCA-50 i.p.- and UDCA-100 i.p.-treated Vil2kd/kd mice (Supplementary Fig. 1). The expression level of IL-1β was not significantly changed in all Vil2kd/kd mice with or without UDCA treatment. These data suggest that UDCA-50 i.p. prevents the progression of periductal fibrosis more effectively in Vil2kd/kd mice by suppressing mRNA expression levels of these inflammatory and profibrotic factors.
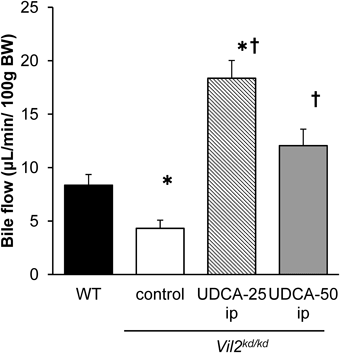
The Effects of Intraperitoneal Administration of UDCA in Bile Flow, Biliary pH and HCO3− Concentration in Vil2kd/kd MiceTo investigate the effect of i.p. administration of UDCA on bile fluidity in Vil2kd/kd mice, we measured bile flow in mice administered with i.p. UDCA (Fig. 5). We found bile flow decreased by about 50% in Vil2kd/kd mice compared with WT mice, similar to as has been previously reported.7) UDCA-25 i.p. and UDCA-50 i.p. treatment significantly enhanced bile flow in Vil2kd/kd mice. These results suggest that i.p. administration of UDCA recovered biliary fluidity in Vil2kd/kd mice. On the other hand, biliary pH and HCO3− concentration were measured in the bile from UDCA-50 i.p.-treated Vil2kd/kd mice. However, biliary pH and HCO3− concentration were not significantly improved in UDCA-50 i.p.-treated Vil2kd/kd mice (Table 4). Both parameters were still similar with those of control Vil2kd/kd mice, suggesting that UDCA improved the liver function independent of improvement of biliary bicarbonate umbrella formation.
Table 4. Biliary pH and HCO
3− Concentration in Mice Administered with UDCA Intraperitoneally for 3 Weeks
| pH | HCO3− (mM) |
|---|
| WT | 8.01±0.02 | 26.7±2.4 |
| Vil2kd/kd |
| Control | 7.71±0.02* | 19.0±1.6* |
| UDCA (50 mg/kg/d) | 7.78±0.07* | 15.1±0.6* |
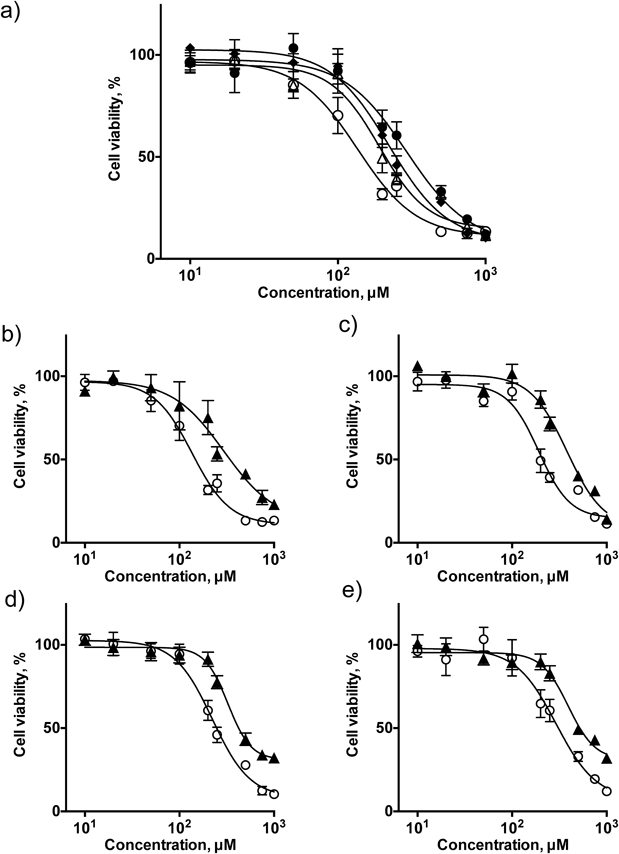
To investigate the mechanism that UDCA protects cholangiocytes from hydrophobic bile acid-induced cytotoxicity under the bicarbonate umbrella dysfunction, we performed in vitro analyses using mouse derived immortalized cholangiocytes. Firstly, we examined a pH dependent cholangiocyte toxicity mediated by a hydrophobic bile acid, chenodeoxycholic acid (CDCA). CDCA exhibited the dose- and pH-dependent cytotoxicity (Fig. 6a). Interestingly, when 100 µM of UDCA was added to the medium, viabilities of NMCs against CDCA-induced cytotoxicity were improved in all pH condition (Fig. 6b) pH 6.5, c) 7.0, d) 7.4, and e) 8.0, respectively), suggesting that UDCA protects cholangiocyte from hydrophobic bile acid-induced cytotoxicity independent of biliary pH.
DISCUSSION
Vil2kd/kd mice showed the characteristic phenotypes of intrahepatic cholestasis including decreased bile fluidity and dysfunction in biliary bicarbonate secretion, as has been previously reported, although we did not observe any lymphocyte infiltration or autoantibody generation.7) Abnormal membrane localization of transporters such as CFTR, AE2, and AQP1, which impairs bicarbonate secretion, is responsible for the progression of intrahepatic cholestasis in Vil2kd/kd mice. Accordingly, these mice are useful for investigating the pharmacological effect of UDCA on cholestatic disease as it relates to impaired biliary bicarbonate umbrella formation.
Previously, Fickert et al. reported that administration of UDCA (0.5% (w/w)) exacerbates the cholestatic phenotypes in Mdr2−/− mice, which resembles the human equivalent of primary sclerosing cholangitis (PSC).14) Mdr2, the rodent orthologue of MDR3, is a member of the ATP-binding cassette transporters, and functions as a canalicular phospholipid flippase. Mutations in MDR3 cause progressive familial intrahepatic cholestasis (PFIC). It has previously been reported that Mdr2−/− mice develop PSC-like liver injury. Furthermore, it has been shown that a diet of 0.5% (w/w) UDCA can be used to observe pharmacological effects in animal experiments.13,14) In Mdr2−/− mice, a 0.5% (w/w) UDCA diet significantly increased plasma AST and ALP concentrations, although cholangiocyte proliferation and periductal fibrosis were slightly improved.14) In our experiments, Vil2kd/kd mice fed a 0.5% (w/w) UDCA diet exhibited similar phenotypes with comparable increases in plasma ALT and AST concentrations as in Mdr2−/− mice.14) On the other hand, our results suggest that lower doses of orally-administered UDCA are appropriate for treating cholestasis in animal models. Furthermore, i.p. injection of lower doses of UDCA significantly improved liver injuries in Vil2kd/kd mice, while administration of 100 mg/kg BW/d of UDCA was shown to increase mortality. The increased mortality may be due to excessive biliary pressure that might result in the rupture of cholangioles and aggravation of hepatocyte necrosis, as was observed in Mdr2−/− mice.14)
Recent studies have also reported that long-term administration of high doses (28–30 mg/kg/d in humans) of UDCA is associated with risk of severe liver dysfunction, and hepatocellular carcinoma in cholestatic patients.17) The toxic side effects of UDCA including inhibition of DNA repair mechanisms, co-enzyme A, cyclic AMP, and NO synthesis have also been reported.1) To prevent such serious adverse events, dosage of UDCA should be strictly considered.
In this study, we found that UDCA is a potent therapeutic agent for the treatment of intrahepatic cholestasis attributed to the functional defect in biliary bicarbonate secretion. It is known that UDCA has multiple mechanisms of action including stabilization of plasma membrane against cytotoxic bile salt-induced cytolysis,18) and prevention of cytotoxic bile salt-induced apoptosis.19) Furthermore, cholehepatic shunting of UDCA will promote the removal of cytotoxic bile from bile acid pool.20) Replacement of cytotoxic hydrophobic bile acids by UDCA in the total bile acid pool would increase the bile fluidity and reduce bile acid-induced liver injury. Furthermore, UDCA-mediated direct stabilization of the plasma membrane of cholangiocytes against hydrophobic bile acids might be important for the therapeutic effects. Hohenester et al. reported that bile acid-induced cholangiocyte toxicity is dependent on the extracellular pH and AE-2-mediated biliary bicarbonate umbrella formation is a key protective mechanism.21) Our results suggest that UDCA show the protective effect independent of the improvement of biliary bicarbonate umbrella formation.
UDCA is now approved for treating PBC, but its efficacy remains a matter of debate.1) The onset of PSC is strongly associated with IBD and UC. In general, bile acids are mainly reabsorbed by the ileum via the apical sodium dependent bile acid transporter (ASBT).3) However, because ASBT expression in patients with IBD or UC patients is reportedly down-regulated,22) oral administration of UDCA might be less effective in PSC patients. We previously performed comprehensive analysis for expressions of membrane transporter in the ileum of Vil2kd/kd mice.16) However, apparent change in ASBT expression was not observed in the ileum of Vil2kd/kd mice, and it is also confirmed by immunostaining (Supplementary Fig. 2). On the other hand, Casaletto et al. previously reported that intestine-specific ezrin conditional knockout (Villin-Cre Ezlox/lox) mice showed increased fat excretion into the feces.23) They suggest that abnormal localization of CFTR lead to the fat malabsorption found in Villin-Cre Ezlox/lox. Bijvelds et al. also revealed that cftr null mice showed similar increased fat excretion and fecal bile salt loss.24) Similar to that observed in the bile ducts, functional defects in CFTR might occur in the intestine. This may lead to intestinal malabsorption of bile acids in Vil2kd/kd mice, as has been reported in cystic fibrosis (CF) patients.25) Because biliary tract lesions found in CF patients resemble those of PSC patients,26) Vil2kd/kd mice might reflect some characteristics seen in PSC patients. Therefore, Vil2kd/kd mouse might be a useful model as the cystic fibrosis related liver disease (CFLD) and PSC. Our results suggest that parenteral feeding of UDCA is effective for treating patients with CFLD and PSC, although high doses of UDCA have strong side effects, and therefore an appropriate dosage should be carefully selected. Although oral administration of UDCA is frequently adopted, Duerksen et al. reported that intravenous administration of UDCA improved total parenteral nutrition (TPN)-induced cholestasis in piglets at about 40 mg/kg BW/d for 3 weeks.27) I.p. injection is often used for small rodents for which intravenous access is challenging, although it is rarely used in humans. Substances injected intraperitoneally are absorbed into the mesenteric vessels and may undergo hepatic metabolism, although some percentage passes directly across the diaphragm through small lacunae into the thoracic lymph, and thus into the circulation.28) Although the pharmacokinetics of substances administered intraperitoneally is similar with those in oral administration, parenteral administration typically shows a higher bioavailability than oral administration. Our results suggest that parenteral administration of UDCA might be more effective for treating intrahepatic cholestasis, particularly in patients with intestinal malabsorption. Furthermore, the appropriate control of bile fluidity, suppression of periductal fibrosis, and cholangiocyte proliferation by UDCA are more appropriate for intrahepatic cholestasis treatment. Because it is difficult to estimate bile flow in vivo and profibrotic gene expression levels in patients, the blood concentration of UDCA should be continuously monitored.
In summary, UDCA administration recovered the hepatic injury in Vil2kd/kd mice independent of biliary bicarbonate secretion, although the dosage and the route of administration of UDCA should be considered when treating cholestatic disease to ensure therapeutic benefit, and to protect patients from severe adverse effects. Moreover, because long-term parenteral administration imposes a heavy burden on patients, development of more effective methods for administering UDCA should be considered.
Acknowledgments
We would like to thank Professor Sachiko Tsukita for providing us Vil2kd/kd mice. We would like to thank Professor Yoshiyuki Ueno for providing us NMC cells. We thank Dr. Yosuke Matsumoto and Dr. Saori Yoshida for their help with breeding and genotyping of the mice. This research was supported in part by the High-Tech Research Center Project for Private Universities. The authors would like to thank Enago (www.enago.jp) for the English language review.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Kotb MA. Molecular mechanisms of ursodeoxycholic acid toxicity & side effects: ursodeoxycholic acid freezes regeneration & induces hibernation mode. Int. J. Mol. Sci., 13, 8882–8914 (2012).
- 2) Oelkers P, Kirby LC, Heubi JE, Dawson PA. Primary bile acid malabsorption caused by mutations in the ileal sodium-dependent bile acid transporter gene (SLC10A2). J. Clin. Invest., 99, 1880–1887 (1997).
- 3) Oelkers P, Dawson PA. Cloning and chromosomal localization of the human ileal lipid-binding protein. Biochim. Biophys. Acta, 1257, 199–202 (1995).
- 4) Dawson PA, Hubbert M, Haywood J, Craddock AL, Zerangue N, Christian WV, Ballatori N. The heteromeric organic solute transporter alpha-beta, Ostalpha-Ostbeta, is an ileal basolateral bile acid transporter. J. Biol. Chem., 280, 6960–6968 (2005).
- 5) Alrefai WA, Gill RK. Bile acid transporters: structure, function, regulation and pathophysiological implications. Pharm. Res., 24, 1803–1823 (2007).
- 6) Levine JS, Burakoff R. Extraintestinal manifestations of inflammatory bowel disease. Gastroenterol. Hepatol. (NY), 7, 235–241 (2011).
- 7) Hatano R, Akiyama K, Tamura A, Hosogi S, Marunaka Y, Caplan MJ, Ueno Y, Tsukita S, Asano S. Knockdown of ezrin causes intrahepatic cholestasis by the dysregulation of bile fluidity in the bile duct epithelium in mice. Hepatology, 61, 1660–1671 (2015).
- 8) Beuers U, Maroni L, Elferink R. The biliary HCO3− umbrella: experimental evidence revisited. Curr. Opin. Gastroenterol., 28, 253–257 (2012).
- 9) Pollheimer MJ, Fickert P. Animal models in primary biliary cirrhosis and primary sclerosing cholangitis. Clin. Rev. Allergy Immunol., 48, 207–217 (2015).
- 10) Salas JT, Banales JM, Sarvide S, Recalde S, Ferrer A, Uriarte I, Oude Elferink RP, Prieto J, Medina JF. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology, 134, 1482–1493 (2008).
- 11) Durie PR, Kent G, Phillips MJ, Ackerley CA. Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am. J. Pathol., 164, 1481–1493 (2004).
- 12) Tamura A, Kikuchi S, Hata M, Katsuno T, Matsui T, Hayashi H, Suzuki Y, Noda T, Tsukita S, Tsukita S. Achlorhydria by ezrin knockdown: defects in the formation/expansion of apical canaliculi in gastric parietal cells. J. Cell Biol., 169, 21–28 (2005).
- 13) Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Pojer C, Zenz R, Lammert F, Stieger B, Meier PJ, Zatloukal K, Denk H, Trauner M. Effects of ursodeoxycholic and cholic acid feeding on hepatocellular transporter expression in mouse liver. Gastroenterology, 121, 170–183 (2001).
- 14) Fickert P, Zollner G, Fuchsbichler A, Stumptner C, Weiglein AH, Lammert F, Marschall HU, Tsybrovskyy O, Zatloukal K, Denk H, Trauner M. Ursodeoxycholic acid aggravates bile infarcts in bile duct-ligated and Mdr2 knockout mice via disruption of cholangioles. Gastroenterology, 123, 1238–1251 (2002).
- 15) Hatano R, Fujii E, Segawa H, Mukaisho K, Matsubara M, Miyamoto K, Hattori T, Sugihara H, Asano S. Ezrin, a membrane cytoskeletal cross-linker, is essential for the regulation of phosphate and calcium homeostasis. Kidney Int., 83, 41–49 (2013).
- 16) Yoshida S, Fukutomi T, Kimura T, Sakurai H, Hatano R, Yamamoto H, Mukaisho K, Hattori T, Sugihara H, Asano S. Comprehensive proteome analysis of brush border membrane fraction of ileum of ezrin knockdown mice. Biomed. Res., 37, 127–139 (2016).
- 17) Lindor KD, Kowdley KV, Luketic VA, Harrison ME, McCashland T, Befeler AS, Harnois D, Jorgensen R, Petz J, Keach J, Mooney J, Sargeant C, Braaten J, Bernard T, King D, Miceli E, Schmoll J, Hoskin T, Thapa P, Enders F. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology, 50, 808–814 (2009).
- 18) Güldütuna S, Deisinger B, Weiss A, Freisleben HJ, Zimmer G, Sipos P, Leuschner U. Ursodeoxycholate stabilizes phospholipid-rich membranes and mimics the effect of cholesterol: investigations on large unilamellar vesicles. Biochim. Biophys. Acta, 1326, 265–274 (1997).
- 19) Rodríguez-Ortigosa CM, Banales JM, Olivas I, Uriarte I, Marín JJ, Corrales FJ, Medina JF, Prieto J. Biliary secretion of S-nitrosoglutathione is involved in the hypercholeresis induced by ursodeoxycholic acid in the normal rat. Hepatology, 52, 667–677 (2010).
- 20) Glaser SS, Alpini G. Activation of the cholehepatic shunt as a potential therapy for primary sclerosing cholangitis. Hepatology, 49, 1795–1797 (2009).
- 21) Hohenester S, Wenniger LM, Paulusma CC, van Vliet SJ, Jefferson DM, Elferink RP, Beuers U. A biliary HCO3− umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology, 55, 173–183 (2012).
- 22) Jahnel J, Fickert P, Hauer A, Högenauer C, Avian A, Trauner M. Inflammatory bowel disease alters intestinal bile acid transporter expression. Drug Metab. Dispos., 42, 1423–1431 (2014).
- 23) Casaletto JB, Saotome I, Curto M, McClatchey AI. Ezrin-mediated apical integrity is required for intestinal homeostasis. Proc. Natl. Acad. Sci. U.S.A., 108, 11924–11929 (2011).
- 24) Bijvelds MJ, Bronsveld I, Havinga R, Sinaasappel M, de Jonge HR, Verkade HJ. Fat absorption in cystic fibrosis mice is impeded by defective lipolysis and post-lipolytic events. Am. J. Physiol. Gastrointest. Liver Physiol., 288, G646–G653 (2005).
- 25) O’Brien S, Mulcahy H, Fenlon H, O’Broin A, Casey M, Burke A, FitzGerald MX, Hegarty JE. Intestinal bile acid malabsorption in cystic fibrosis. Gut, 34, 1137–1141 (1993).
- 26) Sheth S, Shea JC, Bishop MD, Chopra S, Regan MM, Malmberg E, Walker C, Ricci R, Tsui LC, Durie PR, Zielenski J, Freedman SD. Increased prevalence of CFTR mutations and variants and decreased chloride secretion in primary sclerosing cholangitis. Hum. Genet., 113, 286–292 (2003).
- 27) Duerksen DR, Van Aerde JE, Gramlich L, Meddings JB, Chan G, Thomson AB, Clandinin MT. Intravenous ursodeoxycholic acid reduces cholestasis in parenterally fed newborn piglets. Gastroenterology, 111, 1111–1117 (1996).
- 28) Turner PV, Brabb T, Pekow C, Vasbinder MA. Administration of substances to laboratory animals: routes of administration and factors to consider. J. Am. Assoc. Lab. Anim. Sci., 50, 600–613 (2011).