Abstract
The contribution of caspases to hepatic ischemia/reperfusion (I/R)-induced apoptosis has not been completely understood yet. Several studies have demonstrated increased caspase activity during I/R and the protective effect of caspase inhibitors against I/R injuries. However, reports with opposing results also exist. Herein, we examined the contribution of caspases to the I/R-induced hepatic apoptosis in rats using caspase inhibitors and specific substrates of caspases. Hepatic I/R was induced via a 2-h occlusion of the portal vein and the hepatic artery, without conducting bile duct occlusion. DNA laddering and terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate (dUTP) nick end-labeling (TUNEL)-positive cells were increased at 3 h after reperfusion. Pretreatment with caspase inhibitors (Z-Asp-2,6-dichlorobenzoyloxymethylketone (Z-Asp-cmk) 2 or 10 mg/kg intravenously (i.v.), 20 mg/kg intraperitoneally (i.p.), Z-Val-Ala-Asp(OMe)-fluoromethylketone (Z-VAD-fmk) 3 mg/kg i.v.) failed to reduce apoptosis induced by I/R. Interestingly, apoptosis induced by the portal triad (hepatic artery, portal vein, and bile duct) occlusion/reperfusion could be marginally attenuated using Z-Asp-cmk (2 mg/kg i.v.). The cleavage activity for Ac-DEVD-α-(4-methylcoumaryl-7-amide) (MCA), a caspase-3/7/8/9 substrate, was significantly increased by I/R. Conversely, the cleavage activities for Ac-DNLD-MCA and MCA-VDQVDGW[K-DNP]-NH2, specific substrates for caspase-3 and -7 respectively, were decreased by I/R. Protein expression of the cellular inhibitor of apoptosis protein 2 (c-IAP2), an endogenous caspase inhibitor, was increased by ischemia. Nuclear translocation of the apoptosis-inducing factor (AIF), an initiator protein of caspase-independent apoptosis, was also increased during I/R. These results suggest that caspases are inhibited by c-IAP2 induced during ischemia and that AIF may be involved in initiation of apoptosis induced by hepatic I/R without bile duct occlusion.
Hepatic ischemia/reperfusion (I/R) injury is an unavoidable complication of liver surgery, including liver transplantation.1) Apoptotic cell death has been demonstrated during hepatic I/R; thus, inhibition of apoptosis may provide novel therapeutic strategies for attenuating hepatic I/R injury.2–5)
Caspases are a well-known family of proteases playing pivotal roles in apoptotic cell death and inflammation.6) Initiator caspase-8 and -9 activated by various stimuli produce a chain reaction, including activation of executioner caspase-3 and -7, and result in programmed cell death.6) Caspases are regulated by inhibitory as well as stimulatory cellular factors, such as inhibitor of apoptosis proteins (IAPs), endogenous caspase inhibitor proteins.7) Although caspases initiate apoptosis in various types of tissue under diverse stimuli, contribution of caspases to the hepatic I/R-induced apoptotic cell death is still debatable. Several studies have demonstrated the increase in caspase activity during I/R and the protective effect of caspase inhibitors against hepatic I/R injuries.4,5) However, some attempts to highlight any beneficial effects of caspase inhibitors on hepatic I/R injury have been unsuccessful.8)
Furthermore, less-defined apoptotic cell death pathways that do not require caspase activation have been described.9,10) For example, apoptosis inducing factor (AIF) is a key mediator of caspase-independent apoptosis. AIF is a mitochondrion-localized protein and translocates to the nucleus only under some cell death stimuli. However, the contribution of AIF or other caspase-independent pathways in hepatic I/R-induced apoptosis has not yet been demonstrated.
This study aimed to determine the contribution of caspases to the I/R-induced hepatic apoptosis in a rat using caspase inhibitors and the enzymatic analysis of specific fluorogenic substrates of caspases. The contribution of IAPs and AIF to I/R-induced hepatic apoptosis was also investigated.
MATERIALS AND METHODS
Hepatic I/R Injury ModelHepatic I/R procedures were performed as previously described.11) Male Wistar ST rats were obtained from Japan SLC (Hamamatsu, Japan). Per experimental group, 3–6 rats were used. All animal experiments were conducted under the approved guidelines provided by the animal use committee of the Tokushima Bunri University. Briefly, rats weighing 190–250 g were fasted for 12 h prior to surgery. The abdomen was opened under anesthesia with pentobarbital sodium (50 mg/kg, intraperitoneally (i.p.)). The portal vein and hepatic artery of the left and middle lobes were occluded using a vascular clamp. Unless otherwise mentioned, the bile duct was not occluded. The remaining lobes retained an intact blood supply. Two hours after ischemia, the vascular clamp was removed, abdomen was closed, and animals were allowed to awake. Animals were sacrificed at each indicated time after reperfusion. The caspase inhibitors, Z-Asp-2,6-dichlorobenzoyloxymethylketone (Z-Asp-cmk) (2 or 10 mg/kg intravenously (i.v.), 20 mg/kg i.p., Alexis Biochemicals, U.S.A.) or Z-Val-Ala-Asp(OMe)-fluoromethylketone (Z-VAD-fmk) (3 mg/kg i.v., Biomol International, U.S.A.), were administered 2 min before occlusion.
DNA FragmentationLiver tissue was homogenized in Tris-buffered saline (TBS: 10 mM Tris–HCl, 10 mM ethylenediaminetetraacetic acid (EDTA), pH 8.0) and centrifuged (15000×g, 20 min, 4°C). Subsequently, the supernatant was collected. DNA was extracted via the phenol and phenol/chloroform method. It was treated with RNase precipitated overnight in ethanol containing 0.3-M sodium acetate and was finally dissolved in a Tris EDTA (TE) buffer. Samples were electrophoretically separated on a 1.5% agarose gel containing ethidium bromide. DNA was visualized via a UV transilluminator, and the gels were photographed using a CCD camera.
Terminal Deoxynucleotidyl Transferase-Mediated Deoxyuridine Triphosphate (dUTP) Nick-End Labeling (TUNEL) StainingLiver tissue was fixed by phosphate-buffered 15% formaldehyde, embedded in paraffin, and cut into a 3-µm-thick section. TUNEL staining was performed using a commercial kit (Apop tag Peroxidase In Situ Apoptosis Detection Kit, Chemicon, U.S.A.) according to the manufacturer’s instructions.
Caspase ActivitiesLiver tissue was homogenized in sample buffer (25 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), 5 mM EDTA, 0.1% CHAPS, 2 mM dithiothreitol) and was then centrifuged (14000×g, 10 min, 4°C). Subsequently, the supernatant was collected. Samples were diluted in a reaction buffer (20 mM PIPES, 100 mM NaCl, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, 10 mM dithiothreitol) containing specific caspase substrates, namely Ac-DEVD-α-(4-methylcoumaryl-7-amide) (MCA) (caspase-3/7/8/9, Peptide Institute, Osaka, Japan), Ac-DNLD-MCA (caspase-3 specific, Peptide Institute) or MCA-VDQVDGW[K-DNP]-NH2 (caspase-7 specific, Carbiocem, U.S.A.), and were then incubated at 37°C for 60 min. The level of released 7-amino-4-methylcoumarin was measured using a spectrofluorometer with excitation at 360 nm and emission at 465 nm.
Western Blot AnalysisCytosolic protein extract was prepared as follows. Liver tissue was homogenized in ice-cold HEPES buffer [20 mM HEPES-KOH (pH 7.5)], 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 10 mM dithiothreitol, 10 µL/mL protease inhibitor cocktail (SIGMA, U.S.A.). Then, the homogenate was centrifuged (18000×g, 10 min, 4°C) and the supernatant was collected and stored at −80°C until further processing. The mitochondrial6) and nuclear10) extracts were prepared via the previously described methods. Western blot analysis was performed as described previously.10) Anti-cellular inhibitor of apoptosis protein 2 (c-IAP2) antibody, AIF antibody (Santa Cruz Biotechnology, U.S.A.), and anti-cytochrome oxidase subunit I (COI) antibody (Molecular Probes, U.S.A.) were used as the primary antibody.
Statistical AnalysisEach value is expressed as the mean±standard error of the mean (S.E.M.). Statistical differences were evaluated using one-way ANOVA with Bonferroni’s correction. Differences were considered to be significant at p<0.05.
RESULTS
I/R Induces Hepatic ApoptosisOn the DNA fragmentation assay, DNA laddering was induced after reperfusion and peaked at 3 h after reperfusion following 2 h ischemia in the rat liver (Fig. 1A). TUNEL staining also showed the numerous TUNEL-positive cells with nuclear fragmentation 3 h after reperfusion (Fig. 1B).
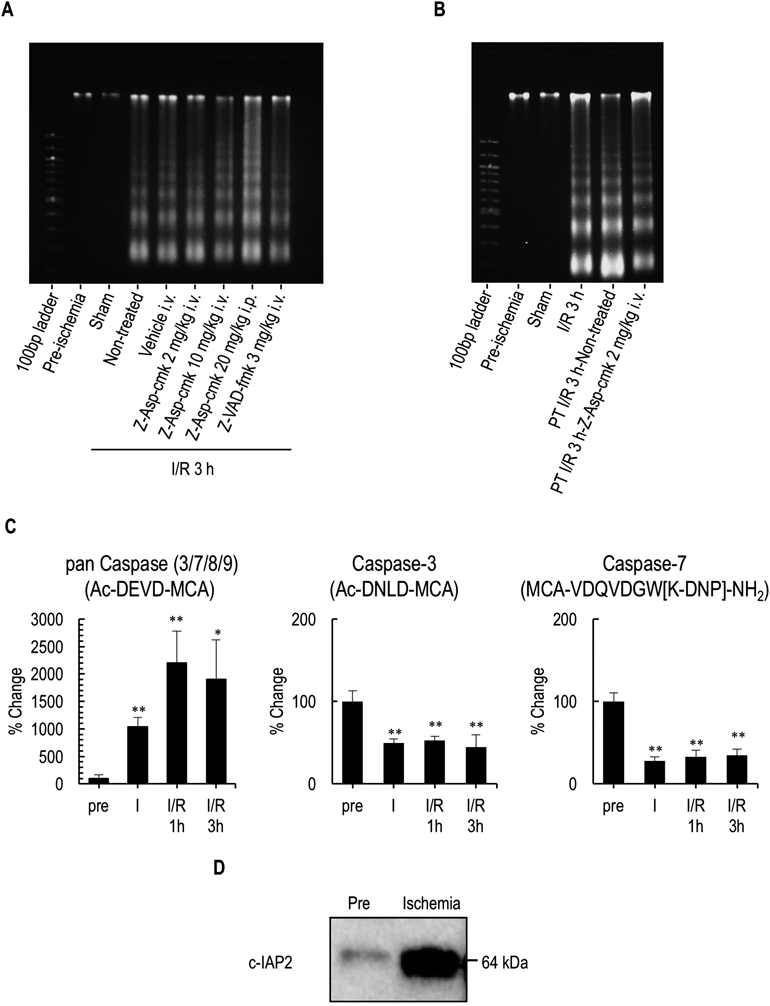
Effects of Caspase Inhibitors on I/R-Induced Hepatic ApoptosisPretreatment with appropriate doses of caspase inhibitors (Z-Asp-cmk 2 or 10 mg/kg i.v., 20 mg/kg i.p., Z-VAD-fmk 3 mg/kg i.v.) did not affect the DNA fragmentation (Fig. 2A) and TUNEL-positive cells (data not shown) 3 h after reperfusion. However, apoptosis induced by the 3-h reperfusion following a 2-h occlusion of the portal triad (PT: hepatic artery, portal vein, and bile duct) was attenuated by pretreatment with Z-Asp-cmk (Fig. 2B).
Caspase Activities in the Liver during I/RCaspase activity was measured enzymatically using specific caspases substrates (Fig. 2C). The cleavage activity for Ac-DEVD-MCA, a pan caspase (caspase-3/7/8/9) substrate, significantly increased following ischemia and subsequent reperfusion. However, the cleavage activities for Ac-DNLD-MCA and MCA-VDQVDGW[K-DNP]-NH2, which are specific substrates for caspase-3 and -7, respectively, were decreased by ischemia and remained low during reperfusion. Western blot analysis showed acute and strong induction in c-IAP2 protein under ischemia (Fig. 2D).
Nuclear Translocation of AIF in the Liver during I/RAIF is a mitochondrion-localized protein and translocates to the nucleus under apoptosis-related stimuli.9) Nuclear translocation of AIF was induced after ischemia and was further enhanced by subsequent reperfusion (Fig. 3). No detectable bands were observed in the Western blot for COI, a mitochondrial marker protein, in the nuclear fraction, indicating that the increase in AIF in the nuclear fraction was not caused by nonspecific contamination of mitochondrial components.
DISCUSSION
The contribution of caspases to hepatic I/R-induced apoptosis remains unclear. Cursio et al. reported that Z-Asp-cmk [0.5 mg/rat (250–300 g) i.v.] reduced I/R-induced TUNEL-positive cells and DNA-fragmentation in the rat hepatic I/R model.4) Soeda et al. reported that Z-VAD-fmk [0.3 mg/rat (200–250 g) i.v.] reduced apoptosis.5) However, similar or higher doses of Z-Asp-cmk and Z-VAD-fmk did not affect the apoptosis induced by I/R in this study. Similarly, Gujral et al. also reported that Z-Asp-cmk did not reduce DNA fragmentation after I/R.8) This is possibly because of the use of different I/R models. Cursio et al. and Soeda et al. used a hepatic I/R model employing portal triad (hepatic artery, portal vein, and bile duct) occlusion. However, similar to Gujral et al., we used a model without bile duct occlusion. This idea is supported by our finding that DNA fragmentation induced by I/R with bile duct occlusion was attenuated by Z-Asp-cmk (Fig. 2B). Moreover, bile duct ligation without ischemia also induced caspase-dependent apoptosis in the liver.12) These results suggest that hepatic apoptosis induced by I/R without bile duct occlusion is caspase inhibitor-resistant.
Executioner caspase-3 and -7 are activated by their upstream cascades, including initiator caspase-8 and -9, and finally lead to proteolytic activation of caspase-activated DNase (CAD). Although Ac-DEVD-MCA has been widely used as a substrate to measure caspase-3 activity,5,8) it is not specific for caspase-3 and is also cleaved by caspase-7, -8, and -9, including initiator caspases.13) Therefore, we used specific substrates for caspase-3 and -7. Cleavage activities for Ac-DNLD-MCA, a specific substrate for caspase-3,13) and MCA-VDQVDGW[K-DNP]-NH2, a specific substrate for caspase-7,14) were decreased during I/R. Conversely, c-IAP2, an endogenous inhibitor of caspase-3, -7 and -9 (but not -8) by binding and degrading via ubiquitination,7) was markedly increased by ischemia in our I/R model. These results suggest that I/R induced initiator caspase-8 activation, but downstream executioner caspase-3 and -7 were inhibited by concurrently activated c-IAP2 in our hepatic I/R model.
AIF is a key mediator of caspase-independent apoptosis. AIF is a mitochondrion-localized protein and translocates to the nucleus by death stimuli.9) In this study, nuclear translocation of AIF was induced by I/R. However, AIF is considered to contribute to large-scale DNA fragmentation (ca. 50 kilobases) but not to low molecular weight DNA fragmentation characterized by DNA laddering. AIF may require cooperative nucleases such as Endonuclease G and L-DNase II for oligonucleosomal DNA cleavage.10) Further studies will be needed to elucidate the mechanism for the I/R-induced caspase-independent apoptosis in liver.
We demonstrated that I/R without bile duct occlusion induced caspase-independent apoptosis in rat liver possibly because of the inhibition of executioner caspase-3 and -7 by c-IAP2 induced by ischemia. We also hypothesize that AIF may partly contribute to I/R-induced caspase-independent hepatic apoptosis. These findings may lead to novel therapeutic strategies to attenuate apoptotic cell death induced by hepatic I/R.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Akhtar MZ, Henderson T, Sutherland A, Vogel T, Friend PJ. Novel approaches to preventing ischemia–reperfusion injury during liver transplantation. Transplant. Proc., 45, 2083–2092 (2013).
- 2) Gao W, Bentley RC, Madden JF, Clavien PA. Apoptosis of sinusoidal endothelial cells is a critical mechanism of preservation injury in rat liver transplantation. Hepatology, 27, 1652–1660 (1998).
- 3) Kohli V, Selzner M, Madden JF, Bentley RC, Clavien PA. Endothelial cell and hepatocyte deaths occur by apoptosis after ischemia–reperfusion injury in the rat liver. Transplantation, 67, 1099–1105 (1999).
- 4) Cursio R, Gugenheim J, Ricci JE, Crenesse D, Rostagno P, Maulon L, Saint-Paul MC, Ferrua B, Auberger AP. A caspase inhibitor fully protects rats against lethal normothermic liver ischemia by inhibition of liver apoptosis. FASEB J., 13, 253–261 (1999).
- 5) Soeda J, Miyagawa S, Sano K, Masumoto J, Taniguchi S, Kawasaki S. Cytochrome c release into cytosol with subsequent caspase activation during warm ischemia in rat liver. Am. J. Physiol. Gastrointest. Liver Physiol., 281, G1115–G1123 (2001).
- 6) McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol., 5, a008656 (2013).
- 7) Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM, Alnemri ES, Salvesen GS, Reed JC. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J., 17, 2215–2223 (1998).
- 8) Gujral JS, Bucci TJ, Farhood A, Jaeschke H. Mechanism of cell death during warm hepatic ischemia–reperfusion in rats: apoptosis or necrosis? Hepatology, 33, 397–405 (2001).
- 9) Daugas E, Nochy D, Ravagnan L, Loeffler M, Susin SA, Zamzami N, Kroemer G. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Lett., 476, 118–123 (2000).
- 10) Samejima K, Earnshaw WC. Trashing the genome: the role of nucleases during apoptosis. Nat. Rev. Mol. Cell Biol., 6, 677–688 (2005).
- 11) Matsui N, Kasajima K, Hada M, Nagata T, Senga N, Yasui Y, Fukuishi N, Akagi M. Inhibition of NF-kappaB activation during ischemia reduces hepatic ischemia/reperfusion injury in rats. J. Toxicol. Sci., 30, 103–110 (2005).
- 12) Sheen-Chen SM, Ho HT, Chen WJ, Eng HL. Effect of ZVAD-fmk on hepatocyte apoptosis after bile duct ligation in rat. World J. Gastroenterol., 11, 2330–2333 (2005).
- 13) Yoshimori A, Sakai J, Sunaga S, Kobayashi T, Takahashi S, Okita N, Takasawa R, Tanuma S. Structural and functional definition of the specificity of a novel caspase-3 inhibitor, Ac-DNLD-CHO. BMC Pharmacol., 7, 8 (2007).
- 14) Talanian RV, Quinlan C, Trautz S, Hackett MC, Mankovich JA, Banach D, Ghayur T, Brady KD, Wong WW. Substrate specificities of caspase family proteases. J. Biol. Chem., 272, 9677–9682 (1997).