Abstract
A significant reduction of glial cell line-derived neurotrophic factor (GDNF) has been identified in the pathophysiology of neurodegenerative and neuropsychiatric disorders. Thus, clarification of the mechanism of GDNF production, and modulating brain GDNF levels could be a novel therapeutic approach. A previous study demonstrated that antidepressant amitriptyline-induced GDNF production was significantly inhibited by pertussis toxin (PTX), a Gi/o protein inhibitor in astrocytes, the main source of GDNF in the brain. However, it is not known whether direct activation of Gi/o protein might induce GDNF expression, and what mechanisms might be involved after Gi/o protein activation. The current study investigated Gi/o protein-initiated GDNF production in rat cortical astrocytes using activators that directly activate Gi/o protein, mastoparan and compound48/80. Treatment of astrocytes with either mastoparan or compound48/80 increased GDNF mRNA expression at 3 and 6 h, and GDNF protein release at 24 h. Treatment of astrocyte with either mastoparan or compound48/80 increased brain-derived neurotrophic factor (BDNF) mRNA expression as well as GDNF. Mastoparan and compound48/80-induced GDNF mRNA expression were significantly inhibited by not only PTX, but also fibroblast growth factor receptor (FGFR) inhibitors, and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase (MEK) inhibitor. In fact, both FGFR substrate2α (FRS2α) and ERK phosphorylation were increased by treatment with either mastoparan or compound48/80, and these were significantly blocked by PTX. Thus, direct, receptor-independent Gi/o protein activation increases GDNF production through FGFR/ERK signaling pathway. The current results indicate a critical role of Gi/o signaling in the regulation of GDNF expression in astrocytes.
Glial cell line-derived neurotrophic factor (GDNF) was originally identified in conditioned medium of a glial cell line as an index of viability of midbrain tyrosine hydroxylase-positive dopaminergic neurons.1) Glial cell line-derived neurotrophic factor is a member of the GDNF family and is broadly expressed in the mammalian brain.2) Glial cell line-derived neurotrophic factor secreted from astrocytes binds to the GDNF-family receptor α1 (GFRα1), which are found on glia, including astrocytes, and neurons, thereby exerting autocrine and paracrine effects.2) The physiological effects of GDNF, including development, differentiation, maintenance and survival of distinct and overlapping neuronal populations within the central nervous system (CNS).2) Additionally, GDNF has been reported to play important roles in higher-order brain functions such as cognition and addiction behavior.3,4)
Clinical studies have shown a reduction in GDNF levels in brain, cerebrospinal fluid and peripheral blood of patients with neurodegenerative and neuropsychiatric disorders, such as Alzheimer’s disease and major depressive disorders.5–7) Preclinical studies in Parkinson’s disease animal models have reported increased survival of nigrostriatal dopaminergic neuron survival following GDNF treatment.8) Thus, GDNF is a potential therapeutic for the treatment of neurological disorders. However, GDNF is a macromolecule that cannot pass the blood–brain barrier. Thus, sufficient, therapeutic levels of brain GDNF cannot be obtained through peripheral administration. Furthermore, the clinical application of exogenous GDNF is limited due to uncertainty of its mechanism of action, short half-life, stability and poor safety profile.5) Stimulating the production of endogenous GDNF could be an alternative approach in increasing brain GDNF levels. Therefore, clarification of the intracellular mechanism involved in the production of GDNF is needed in order to develop a potentially novel therapeutic approach for neurodegenerative and neuropsychiatric disorders.
Previous studies have demonstrated that antiparkinsonian drugs and antidepressant drugs increase GDNF production in astrocytes.9–11) The tricyclic antidepressant amitriptyline increased GDNF production through a pertussis toxin (PTX) sensitive Gi/o protein signaling, which is entirely independent of monoamines.12) An electrical impedance-based biosensor (CellKey™) assay showed that amitriptyline actually activated Gi/o protein signaling in astrocytes.12) Thus, although these findings demonstrate that the Gi/o protein is a key molecule regulating GDNF expression, it is not known whether direct activation of Gi/o protein could induce GDNF expression, and what mechanism might be involved after Gi/o protein activation.
The current study utilized a pharmacological approach to determine if direct activation of Gi/o protein leads to increased GDNF production, and examined the effect of Gi/o protein activator on brain-derived neurotrophic factor (BDNF), vascular endothelial growth factor (VEGF), and fibroblast growth factor 2 (FGF2) mRNA expressions which are increased by amitriptyline treatment.13) Primary cultured rat cortical astrocytes were used because astrocytes are the main source of neurotrophic factors including GDNF in the brain. In addition, we previously found that antidepressants do not evoke GDNF expression in neurons.13) Receptor-bypassing Gi/o protein activators, mastoparan, a peptide isolated from wasp venom, and compound48/80, a synthetic basic secretagogue were utilized.14–17) Additionally, pharmacological tools were used to delineate the cellular signaling mechanism involved in the Gi/o protein-initiated expression of GDNF mRNA. The current study focused on fibroblast growth factor receptor (FGFR) and extracellular signal-regulated kinase (ERK) because FGFR/ERK signaling has been shown to play an important role on GDNF production.18) Understanding the mechanism of GDNF production at the cellular level could lead to the development of a novel approach for treating neurodegenerative and neuropsychiatric disorders.
MATERIALS AND METHODS
All experimental procedures were performed according to both the “Guiding Principles for the Care and Use of Laboratory Animals,” approved by the Japanese Pharmacological Society, and the guidelines for experimental animal care and use of Hiroshima University. The present study was reviewed and approved by the Animal Ethics Committee of Hiroshima University (Approval number: A16-127).
MaterialsGi/o protein activators mastoparan and mastopran-7 were obtained from Peptide Institute, Inc. (Osaka, Japan) and compound48/80 was obtained from Sigma-Aldrich Co. (St. Louis, MO, U.S.A.). Pertussis toxin, and U0126 were obtained from Calbiochem (San Diego, CA, U.S.A.). SU5402, and PD173074 were obtained from Merck KGaA (Darmstadt, Germany). The concentrations of inhibitors (PTX, U0126, SU5402, and PD173074) used in this study selectively inhibit its target, and no other kinases, in rat primary cultured astrocytes.12,19) U0126, SU5402, and PD173074 were dissolved in dimethyl sulfoxide (DMSO). The final concentration of DMSO was adjusted to 0.1% for drug treatment experiments.
Cell Culture and Drug TreatmentPrimary cultured astrocytes were prepared from 1-d-old neonatal Wistar rats, as described previously.19,20) Astrocytes were plated onto 35 mm diameter dishes (1×106 cells/dish). After 2 d, the medium was replaced with serum-free Dulbecco’s modified Eagle’s medium (DMEM). The cells were used for experiments on the following day.
Potential cytotoxicity of mastoparan and compound48/80 in astrocyte was evaluated by the lactate dehydrogenase (LDH) assay and the tetrazolium salt 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich Co.) assay as described previously.13) Cells incubated with either mastoparan or compound48/80 treatment at concentrations used in the current study did not significantly increase media levels of LDH (Supplementary Fig. 1A). Also, cell viability was not affected by concentrations of mastoparan and compound48/80 as determined by the MTT assay (Supplementary Fig. 1B).
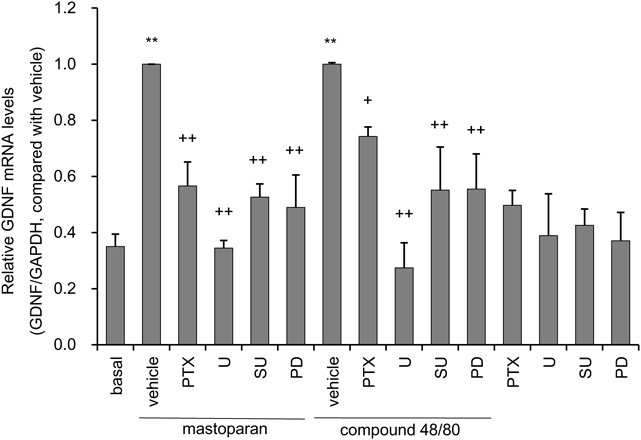
It is possible that mastoparan and compound48/80 interact with other signaling molecules, in addition to Gi/o protein, due to their amphiphilic properties.21,22) To confirm that the effects of mastoparan and compound48/80 on GDNF expression were mediated through Gi/o proteins, astrocytes were incubated with the Gi/o protein inhibitor PTX (100 ng/mL). Several intracellular signaling molecules, including FGFR and ERK, contribute to the regulation of GDNF mRNA expression in astrocytes.18) To determine if FGFR, and ERK are involved in the mastoparan and compound48/80-induced expression of GDNF mRNA, astrocytes were incubated with SU5402 (25 µM) and PD173074 (1 µM), FGFR inhibitors and U0126 (10 µM), a MEK inhibitor. Astrocytes were pre-treated either with or without inhibitors for 0.5 h, and subsequently treated with Gi/o protein activators for 3 h.
Total RNA in astrocytes was prepared by a previously described method,23) and used to synthesize cDNA with MuLV reverse transcriptase (Applied Biosystems, Forster City, CA, U.S.A.) and a random hexamer primer.
Real-Time PCR AnalysiscDNA synthesized using 1 µg of total RNA in each sample were subjected to real-time PCR assays with specific primers and EXPRESS SYBR® GreenER™ qPCR SuperMixes (Invitrogen, Carlsbad, CA, U.S.A.). The expression of GDNF, BDNF, VEGF, and FGF2 mRNA were detected by using a pair of primer.13) Real-time PCR assays were conducted using a DNA engine Opticon 2 real-time PCR detection system (Bio-Rad Laboratories, Inc., Hercules, CA, U.S.A.). The three-step amplification protocol consisted of 3 min at 95°C, followed by 40 cycles at 95°C for 15 s, 60°C for 30 s, and 72°C for 30s. RNA quantities of target genes were calculated using the Ct method. The Ct values of GDNF, BDNF, VEGF, and FGF2 amplification was normalized to that of glyceraldehydes-3-phosphate dehydrogenase (GAPDH) amplification.
Enzyme-Linked Immunosorbent Assay (ELISA)For the GDNF release assay, astrocytes were cultured at a density of 0.5×106 cells on a 12-well plate with 1 mL of medium. After drug treatment, the concentration of GDNF protein in cell-conditioned media was determined using a GDNF Emax® ImmunoAssay System (Promega, Madison, WI, U.S.A.) according to the manufacturer’s instructions. The percent recovery of GDNF (31–1000 pg/mL) added to astrocyte conditioning medium was between 88–126%.
Western BlottingThe following antibodies were used: phospho-FGFR substrate 2α (FRS2α) (Tyr196) antibody, p44/42 MAPK (ERK1/2), and phospho-p44/42 MAPK (ERK1/2) (Cell Signaling Technology, Danvers, MA, U.S.A.); and FRS2α (SNT-1) antibody (Sigma-Aldrich Co.). Briefly, the cell lysis proteins were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and transblotted to polyvinylidene difluoride membranes. The membranes were blocked with 5% (w/v) bovine serum albumin (BSA) or skim milk for 6 h at 4°C and incubated with respective antibodies overnight at 4°C. After washing, the membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Chemiluminescent detection was performed using Immun-Star WesternC™ kit (Bio-Rad), and the net intensities of each band were quantified using GE Healthcare Life Sciences Image Quant LAS 4000 (Waukesha, WI, U.S.A.).
Statistical AnalysisData are expressed as the mean±standard error of the mean (S.E.M.) of at least three independent experiments. Statistically significant differences among more than two treatment groups were determined using one-way ANOVA with pairwise comparison carried out by either Tukey’s honest significant difference (HSD) or Dunnett’s test. Statistically significant differences between two treatment groups were analyzed by Student’s t-test. p values at less than 0.05 were taken as statistically significant.
RESULTS
Effects of Gi/o Protein Activators on GDNF Production in Rat Primary Cultured Cortical AstrocytesMastoparan, at 5 µM, but not 0.1 or 1 µM, significantly increased astrocytic GDNF mRNA expression (Fig. 1A). Expression of GDNF mRNA significantly increased 3 h after the beginning of mastoparan treatment and expression was sustained 6 h after the beginning of treatment (Fig. 1B). Mastoparan-7 is a potent analogue of the peptide mastoparan, and the EC50 of mastoparan-7 is 5-fold lower than that of mastoparan in GTPase and GDP exchange assays which indicate Gi/o protein activation.15) For example, in a rat vascular smooth muscle assay, the EC50 of mastoparan-7 is 44.1±2.3 nM, which is rightward shifted in the presence of PTX,24) confirming in a different cell type an involvement of PTX-sensitive Gi/o protein. Mastoparan-7 also significantly increased GDNF mRNA expression (Fig. 1A). Compound48/80 also increased GDNF mRNA expression in a concentration-dependent manner and a statistically significant increase in expression was observed at concentrations above 10 µg/mL (Fig. 1C). Similar to mastoparan and mastoparan-7, compound48/80 significantly increased GDNF mRNA at 3 h and 6 h after the beginning of treatment (Fig. 1D). Furthermore, treatment with mastoparan and compound48/80 for 24 h significantly increased GDNF protein release as measured in the medium (Table 1).
Table 1. Effects of Gi/o Protein Activators on GDNF Release in Rat Primary Cultured Cortical Astrocytes
| GDNF released (pg/mL) |
|---|
| Basal | 10.5±2.3 |
| Mastoparan (5 µM) | 21.2±4.4* |
| Compound48/80 (10 µg/mL) | 33.1±2.2** |
Astrocytes were treated with mastoparan or compound48/80 for 24 h, and GDNF released into the medium was analyzed by ELISA. The data are expressed as the mean±S.E.M. (pg/mL) * p<0.05, ** p<0.01 vs. basal (Student’s t-test; n=5).
Treatment with either mastoparan or compound48/80 significantly increased BDNF mRNA expression as well as GDNF. However, the expression of VEGF mRNA was decreased by treatment with either mastoparan or compound48/80. Compound48/80 increased FGF2 mRNA expression, while mastoparan did not have effect (Table 2).
Table 2. Effects of Gi/o Protein Activators on Neurotrophic/Growth Factors mRNA Expression in Rat Primary Cultured Cortical Astrocytes
| BDNF mRNA (Ratio of basal) | VEGF mRNA (Ratio of basal) | FGF2 mRNA (Ratio of basal) |
|---|
| Mastoparan (5 µM) | 4.06±1.21* | 0.61±0.13* | 1.02±0.13 |
| Compound48/80 (10 µg/mL) | 13.03±4.95* | 0.47±0.18* | 1.51±0.16* |
Astrocytes were treated with mastoparan or compound48/80 for 3 h, and BDNF, VEGF, or FGF2 mRNA expression levels were analyzed by real-time PCR. The values are shown as the ration of BDNF, VEGF, or FGF2 mRNA to GAPDH mRNA. The data are expressed as the mean±S.E.M. * p<0.05 vs. basal (Student’s t-test; n=4–7).
Both mastoparan and compound48/80-induced expression of GDNF mRNA were significantly inhibited by PTX. Pretreatment with MEK inhibitor U0126 suppressed most of the effect of mastoparan and compound48/80 on astrocytic GDNF mRNA expression. Mastoparan and compound48/80-evoked GDNF mRNA expression were significantly blocked by FGFR inhibitors SU5402 and PD173074 (Fig. 2). Treatment with these inhibitors alone did not affect GDNF mRNA expression.
Effects of Gi/o Protein Activators on Both FRS2α and ERK Phosphorylation in Rat Primary Cultured Cortical AstrocytesTreatment with either mastoparan or compound48/80 significantly increased phosphorylation of FRS2α, a surrogate of FGFR activation (Figs. 3A, B), and phosphorylation of ERK (Figs. 3C, D). The mastoparan or compound48/80-induced FRS2α phosphorylation and ERK phosphorylation were significantly inhibited by pretreatment of PTX (Fig. 3). Treatment with PTX alone did not affect basal level of FRS2α phosphorylation and ERK phosphorylation.
DISCUSSION
The current study demonstrated that direct, receptor-independent Gi/o protein activation increased rat brain astrocytic GDNF mRNA expression and GDNF release. Furthermore, Gi/o protein activation initiated intracellular signaling though FGFR and ERK, which appear to be necessary for GDNF mRNA expression. The current findings in total indicate that Gi/o signaling is crucial in regulating GDNF production in brain astrocytes.
Astrocytes express several types of alpha subunit of G protein, including Gi/o, Gs, and Gq, and these have been shown to be involved in intercellular signaling in response to various chemical stimuli in vitro.25,26) A previous study showed that amitriptyline induced GDNF production in astrocytes, which was blocked by PTX, but not YM254890 (Gq inhibitor), or NF449 (Gs inhibitor).12) Both amitriptyline-induced Gi/o protein activation and GDNF production were blocked by lysophosphatidic acid (LPA) receptor 1 antagonists,27) indicating that the Gi/o protein-coupled LPA receptor 1 could be involved in the cellular effect of amitriptyline. These results suggest that the Gi/o protein is crucial in regulation of GDNF expression. However, it is not entirely clear whether Gi/o protein activation is pivotal in GDNF production. The Gi/o protein is broadly expressed in CNS,28) and mediates various intracellular signaling processes. In the current study, direct, receptor-independent Gi/o protein activation increased GDNF production, and the efficacy of Gi/o protein activators to induce GDNF expression appears to be much greater than that of amitriptyline.13) Thus, the potency of Gi/o protein activation reflected the amount of GDNF production. Furthermore, Gi/o protein activators have been shown to increase not only GDNF expression, but also BDNF, which has been shown to be increased by amitriptyline, and important for the therapeutic effect of antidepressants as well as GDNF.29) Thus, potentiating Gi/o protein signaling efficacy of amitriptyline, in order to boost endogenous GDNF and BDNF production, could be a highly effective therapeutic strategy.
FGFR and ERK are involved in amitriptyline-induced expression of GDNF mRNA in astrocytes.18) However, it is unknown whether these intracellular signaling molecules contribute to Gi/o protein activator-mediated GDNF expression. The current study suggests that FGFR and ERK do have roles in Gi/o protein activator-mediated GDNF expression as inhibitors to these molecules significantly attenuated mastoparan and compound48/80-induced GDNF mRNA expression. Furthermore, the current study shows that treatment with either mastoparan or compound48/80 significantly increases phosphorylation of FRS2α and ERK, which were blocked by pretreatment of PTX. Thus, these results suggest that FGFR and ERK are crucial for Gi/o protein-mediated GDNF mRNA expression.
The effects of the Gi/o protein activators were in fact mediated through the Gi/o protein as the effects were blocked by Gi/o protein inhibitor PTX. In the current study, mastoparan and compound48/80-induced GDNF mRNA expression were partially, albeit statistically significant, inhibited with PTX pretreatment. FRS2α phosphorylation and ERK phosphorylation by either mastoparan or compound48/80 were also significantly inhibited by PTX. An additional mechanism, other than a Gi/o protein/FGFR/ERK cascade, could be involved in compound48/80-induced GDNF mRNA expression, since PTX almost completely inhibited FRS2α and ERK1/2 phosphorylation but only partially blocked GDNF mRNA expression. Multiple signaling pathways, including protein kinase C (PKC) and Ca2+, are involved in the regulation in GDNF production.30) Compound48/80 demonstrated an effect on PKC and Ca2+ signaling in addition to Gi/o protein activation,22) thus these signaling pathways could be involved in the compound48/80-induced GDNF mRNA expression. The concentrations of mastoparan and compound48/80 needed to activate Gi/o protein in the current study are close to the concentrations that lead to enhanced membrane permeability.31) Thus, the current study conducted an LDH release assay to determine if increased GDNF expression was not merely due to increased membrane permeability. In addition, the MTT cell viability assay demonstrated that GDNF production was not a result of nonspecific cytotoxicity.
The Gi/o protein activators increase both GDNF and BDNF mRNA expression in astrocytes. However, mastoparan and compound48/80 treatment decreased VEGF mRNA expression. Thus, expression of these neurotrophic factors in astrocytes could be differentially regulated by Gi/o protein signaling. Both mastoparan and compound48/80 increased GDNF mRNA expression to a similar degree, but compound48/80 treatment increased BDNF mRNA to a greater extent than mastoparan treatment. Treatment with compound48/80, but not mastoparan, increased FGF2 mRNA expression. Compound48/80 activates a number of intracellular signaling pathways in addition to Gi/o activation.32) Thus, while both compound48/80 and mastoparan activate intracellular Gi/o signaling, other intracellular mechanisms could underlie the effect of compound48/80 on neurotrophic factor expression in the current study.
Mastoparan was first identified and chemically characterized by Hirai et al. in 1979,33) and a subsequent study demonstrated that mastoparan has a direct Gi/o protein activating potency.14) Natural, modified and chimeric analogs of mastoparan, which are potent Gi/o protein activators, have been used to treat a number of neurological conditions.34) In fact, mastoparan-7 demonstrates significant biological effects such as dendritic spine remodeling, increasing spine numbers, and recruitment of postsynaptic density protein 95 (PSD-95) into spines to produce functional synapses in mature hippocampal neurons.35) GDNF and BDNF signaling regulate synaptic protein synthesis and synapse formation.36,37) Thus, the biological effects of mastoparan-7 could be mediated by increases in endogenous GDNF and BDNF production following Gi/o protein signaling activation. Decreased synaptic efficacy and impaired neural functioning have been suggested as underlying causes of neuropsychiatric and neurodegenerative disorders.38) Enhancement of endogenous neurotrophic factor, such as GDNF and BDNF could be beneficial in promoting recovery from these disorders. Previous in vivo studies have demonstrated behavioral response to antidepressants involves increasing GDNF production and PTX-sensitive signaling in the brain.39,40) The potentiating Gi/o protein signaling efficacy of antidepressant by combination treatment with Gi/o protein activator could boost endogenous GDNF and BDNF production. This would suggest a potential mechanism that could be targeted in the development of novel antidepressant treatments. A dysfunctional PTX-sensitive Gi/o system could be a key intracellular mechanism that underlies patients with affective disorders.41) In fact, GDNF expression is decreased in these patients5) and may suggest a change in the intracellular mechanism that mediates GDNF expression. It is also possible that this change in the PTX-sensitive Gi/o system could in part explain the lack of efficacy of antidepressant treatment in some patients.
While mastoparan and compound48/80 increase GDNF expression, the compounds themselves would not be therapeutically useful since these compounds activated other intracellular pathways in addition to Gi/o protein activation. Thus, selective Gi/o protein activators or modification of existing antidepressants to potentiate the efficacy of Gi/o activation could lead to new antidepressant drugs.
Acknowledgments
This work was supported in part by the Japan Society for the Promotion of Science (JSPS) KAKENHI Grant Number JP16K082700, and by Grants from the Naito Foundation. We wish to thank the Analysis Center of Life Science and the Natural Science Center for Basic Research and Development, Hiroshima University for the use of their facilities. We also thank Dr. Aldric T. Hama for his careful editing of the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science, 260, 1130–1132 (1993).
- 2) Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat. Rev. Neurosci., 3, 383–394 (2002).
- 3) Messer CJ, Eisch AJ, Carlezon WA Jr, Whisler K, Shen L, Wolf DH, Westphal H, Collins F, Russell DS, Nestler EJ. Role for GDNF in biochemical and behavioral adaptations to drugs of abuse. Neuron, 26, 247–257 (2000).
- 4) Gerlai R, McNamara A, Choi-Lundberg DL, Armanini M, Ross J, Powell-Braxton L, Phillips HS. Impaired water maze learning performance without altered dopaminergic function in mice heterozygous for the GDNF mutation. Eur. J. Neurosci., 14, 1153–1163 (2001).
- 5) Ibáñez CF, Andressoo JO. Biology of GDNF and its receptors—relevance for disorders of the central nervous system. Neurobiol. Dis., 97 (Part B), 80–89 (2017).
- 6) Sharma AN, da Costa e Silva BF, Soares JC, Carvalho AF, Quevedo J. Role of trophic factors GDNF, IGF-1 and VEGF in major depressive disorder: a comprehensive review of human studies. J. Affect. Disord., 197, 9–20 (2016).
- 7) Budni J, Bellettini-Santos T, Mina F, Garcez ML, Zugno AI. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis., 6, 331–341 (2015).
- 8) d’Anglemont de Tassigny X, Pascual A, López-Barneo J. GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease. Front Neuroanat., 9, 10 (2015).
- 9) Ohta M, Mizuta I, Ohta K, Nishimura M, Mizuta E, Hayashi K, Kuno S. Apomorphine up-regulates NGF and GDNF synthesis in cultured mouse astrocytes. Biochem. Biophys. Res. Commun., 272, 18–22 (2000).
- 10) Mizuta I, Ohta M, Ohta K, Nishimura M, Mizuta E, Hayashi K, Kuno S. Selegiline and desmethylselegiline stimulate NGF, BDNF, and GDNF synthesis in cultured mouse astrocytes. Biochem. Biophys. Res. Commun., 279, 751–755 (2000).
- 11) Hisaoka K, Nishida A, Koda T, Miyata M, Zensho H, Morinobu S, Ohta M, Yamawaki S. Antidepressant drug treatments induce glial cell line-derived neurotrophic factor (GDNF) synthesis and release in rat C6 glioblastoma cells. J. Neurochem., 79, 25–34 (2001).
- 12) Hisaoka-Nakashima K, Miyano K, Matsumoto C, Kajitani N, Abe H, Okada-Tsuchioka M, Yokoyama A, Uezono Y, Morioka N, Nakata Y, Takebayashi M. Tricyclic antidepressant amitriptyline-induced glial cell line-derived neurotrophic factor production involves pertussis toxin-sensitive Gαi/o activation in astroglial cells. J. Biol. Chem., 290, 13678–13691 (2015).
- 13) Kajitani N, Hisaoka-Nakashima K, Morioka N, Okada-Tsuchioka M, Kaneko M, Kasai M, Shibasaki C, Nakata Y, Takebayashi M. Antidepressant acts on astrocytes leading to an increase in the expression of neurotrophic/growth factors: differential regulation of FGF-2 by noradrenaline. PLOS ONE, 7, e51197 (2012).
- 14) Higashijima T, Uzu S, Nakajima T, Ross EM. Mastoparan, a peptide toxin from wasp venom, mimics receptors by activating GTP-binding regulatory proteins (G proteins). J. Biol. Chem., 263, 6491–6494 (1988).
- 15) Higashijima T, Burnier J, Ross EM. Regulation of Gi and Go by mastoparan, related amphiphilic peptides, and hydrophobic amines. Mechanism and structural determinants of activity. J. Biol. Chem., 265, 14176–14186 (1990).
- 16) Mousli M, Bronner C, Bockaert J, Rouot B, Landry Y. Interaction of substance P, compound48/80 and mastoparan with the alpha-subunit C-terminus of G protein. Immunol. Lett., 25, 355–357 (1990).
- 17) Mousli M, Bronner C, Landry Y, Bockaert J, Rouot B. Direct activation of GTP-binding regulatory proteins (G-proteins) by substance P and compound 48/80. FEBS Lett., 259, 260–262 (1990).
- 18) Hisaoka K, Tsuchioka M, Yano R, Maeda N, Kajitani N, Morioka N, Nakata Y, Takebayashi M. Tricyclic antidepressant amitriptyline activates fibroblast growth factor receptor signaling in glial cells: involvement in glial cell line-derived neurotrophic factor production. J. Biol. Chem., 286, 21118–21128 (2011).
- 19) Kajitani N, Hisaoka-Nakashima K, Okada-Tsuchioka M, Hosoi M, Yokoe T, Morioka N, Nakata Y, Takebayashi M. Fibroblast growth factor 2 mRNA expression evoked by amitriptyline involves extracellular signal-regulated kinase-dependent early growth response 1 production in rat primary cultured astrocytes. J. Neurochem., 135, 27–37 (2015).
- 20) Morioka N, Suekama K, Zhang FF, Kajitani N, Hisaoka-Nakashima K, Takebayashi M, Nakata Y. Amitriptyline up-regulates connexin43-gap junction in rat cultured cortical astrocytes via activation of the p38 and c-Fos/AP-1 signalling pathway. Br. J. Pharmacol., 171, 2854–2867 (2014).
- 21) Nakahata N, Sugama J. Pharmacological activity of mastoparan: its contribution to signal transduction. Nippon Yakurigaku Zasshi, 136, 145–149 (2010).
- 22) Shefler I, Seger R, Sagi-Eisenberg R. Gi-mediated activation of mitogen-activated protein kinase (MAPK) pathway by receptor mimetic basic secretagogues of connective tissue-type mast cells: bifurcation of arachidonic acid-induced release upstream of MAPK. J. Pharmacol. Exp. Ther., 289, 1654–1661 (1999).
- 23) Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal. Biochem., 162, 156–159 (1987).
- 24) Grześk G, Malinowski B, Grześk E, Wiciński M, Szadujkis-Szadurska K. Direct regulation of vascular smooth muscle contraction by mastoparan-7. Biomed. Rep., 2, 34–38 (2014).
- 25) Hamby ME, Coppola G, Ao Y, Geschwind DH, Khakh BS, Sofroniew MV. Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J. Neurosci., 32, 14489–14510 (2012).
- 26) Viollet C, Loudes C, Kordon C, Faivre-Bauman A. Selective patterns of expression of G protein alpha subunits during in vitro development of hypothalamic neurons. J. Neurochem., 63, 2231–2239 (1994).
- 27) Kajitani N, Miyano K, Okada-Tsuchioka M, Abe H, Itagaki K, Hisaoka-Nakashima K, Morioka N, Uezono Y, Takebayashi M. Identification of lysophosphatidic acid receptor 1 in astroglial cells as a target for glial cell line-derived neurotrophic factor expression induced by antidepressants. J. Biol. Chem., 291, 27364–27370 (2016).
- 28) Milligan G, Kostenis E. Heterotrimeric G-proteins: a short history. Br. J. Pharmacol., 147 (Suppl. 1), S46–S55 (2006).
- 29) Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav. Pharmacol., 18, 391–418 (2007).
- 30) Verity AN, Wyatt TL, Hajos B, Eglen RM, Baecker PA, Johnson RM. Regulation of glial cell line-derived neurotrophic factor release from rat C6 glioblastoma cells. J. Neurochem., 70, 531–539 (1998).
- 31) Tanimura A, Matsumoto Y, Tojyo Y. Mastoparan increases membrane permeability in rat parotid cells independently of action on G-proteins. Biochem. Biophys. Res. Commun., 177, 802–808 (1991).
- 32) Palomäki VA, Laitinen JT. The basic secretagogue compound 48/80 activates G proteins indirectly via stimulation of phospholipase D-lysophosphatidic acid receptor axis and 5-HT1A receptors in rat brain sections. Br. J. Pharmacol., 147, 596–606 (2006).
- 33) Hirai Y, Yasuhara T, Yoshida H, Nakajima T, Fujino M, Kitada C. A new mast cell degranulating peptide “mastoparan” in the venom of Vespula lewisii. Chem. Pharm. Bull., 27, 1942–1944 (1979).
- 34) Silva J, Monge-Fuentes V, Gomes F, Lopes K, dos Anjos L, Campos G, Arenas C, Biolchi A, Gonçalves J, Galante P, Campos L, Mortari M. Pharmacological alternatives for the treatment of neurodegenerative disorders: wasp and bee venoms and their components as new neuroactive tools. Toxins (Basel), 7, 3179–3209 (2015).
- 35) Ramírez VT, Ramos-Fernández E, Inestrosa NC. The Gαo activator mastoparan-7 promotes dendritic spine formation in hippocampal neurons. Neural Plast., 2016, 4258171 (2016).
- 36) Duman CH, Duman RS. Spine synapse remodeling in the pathophysiology and treatment of depression. Neurosci. Lett., 601, 20–29 (2015).
- 37) Ledda F, Paratcha G, Sandoval-Guzmán T, Ibáñez CF. GDNF and GFRalpha1 promote formation of neuronal synapses by ligand-induced cell adhesion. Nat. Neurosci., 10, 293–300 (2007).
- 38) Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science, 338, 68–72 (2012).
- 39) Uchida S, Hara K, Kobayashi A, Otsuki K, Yamagata H, Hobara T, Suzuki T, Miyata N, Watanabe Y. Epigenetic status of GDNF in the ventral striatum determines susceptibility and adaptation to daily stressful events. Neuron, 69, 359–372 (2011).
- 40) Galeotti N, Bartolini A, Ghelardini C. Role of Gi proteins in the antidepressant-like effect of amitriptyline and clomipramine. Neuropsychopharmacology, 27, 554–564 (2002).
- 41) Grammatopoulos DK. Regulation of G-protein coupled receptor signalling underpinning neurobiology of mood disorders and depression. Mol. Cell. Endocrinol., 449, 82–89 (2017).