Abstract
Law enforcement against illicit use of cannabis and related substances requires rapid, feasible, and reliable tools for on-site testing of cannabinoids. Notably, methods based on cannabinoid-specific antibodies enable efficient screening of multiple specimens. Antibody engineering may accelerate development of modern and robust testing systems. Here, we used in vitro affinity maturation to generate a single-chain Fv fragment (scFv) that recognizes with high affinity the psychoactive cannabinoid, Δ9-tetrahydrocannabinol (THC). A mouse monoclonal antibody against THC, Ab-THC#33, with Ka 6.2×107 M−1 (as Fab fragment) was established by the hybridoma technique. Then, a “wild-type” scFv (wt-scFv) with Ka, 1.1×107 M−1 was prepared by bacterial expression of a fusion gene combining the VH and VL genes for Ab-THC#33. Subsequently, random point mutations in VH and VL were generated separately, and the resulting products were assembled into mutant scFv genes, which were then phage-displayed. Repeated panning identified a mutant scFv (scFv#m1-36) with 10-fold enhanced affinity (Ka 1.1×108 M−1) for THC, in which only a single conservative substitution (Ser50Thr) was present at the N-terminus of the VH-complementarity-determining region 2 (CDR2) sequence. In competitive enzyme-linked immunosorbent assay (ELISA), the mutant scFv generated dose–response curves with midpoint 0.27 ng/assay THC, which was 3-fold lower than that of wt-scFv. Even higher reactivity with a major THC metabolite, 11-nor-9-carboxy-Δ9-tetrahydrocannabinol, indicated that the mutant scFv will be useful for testing not only THC in confiscated materials, but also the metabolite in urine. Indeed, the antibody fragment is potentially suitable for use in advanced on-site testing platforms for cannabinoids.
Cannabis, i.e., marijuana, is the crude drug derived from the plant Cannabis sativa L., and has long been one of the most widely abused illicit substances in the world.1–5) A previous report demonstrated that cannabis contains more than 500 compounds, 109 of which are cannabinoids, a class of compounds that act on cannabinoid receptors in cells.2) Of these cannabinoids, Δ9-tetrahydrocannabinol (THC) is the primary psychoactive compound.1–5) However, the most abundant compound accounting for ca. 95% of cannabinoids in fresh, dried marijuana, is the non-psychoactive compound, Δ9-tetrahydrocannabinolic acid (THCA),4) which is easily decarboxylated to THC by drying or heating (Fig. 1A). In turn, THC decomposes to cannabinol (CBN) by exposure to light and oxygen1) (Fig. 1A). In humans, THC is biotransformed mainly according to Fig. 1B, resulting in more polar metabolites, including 11-hydroxy-Δ9-tetrahydrocannabinol, 11-nor-9-carboxy-Δ9-tetrahydrocannabinol (THC-COOH), and its conjugate with glucuronic acid (THC-COOGlu).5,6)
Advanced on-site testing that enables quick and convenient detection of THC and related compounds will facilitate crackdowns on illicit use of cannabis. Antibody-based tests are more suitable for this purpose than chromatographic methods, which require expensive and fixed instruments as well as time-consuming sample pretreatment steps. For example, lateral-flow immunoassays, i.e., immunochromatographies, are already available for on-site testing of illicit drugs, including THC and its metabolites.7)
Genetically engineering of antibodies (antibody engineering) has also generated artificial antibodies with valuable functions.8,9) For instance, native antibodies (immunoglobulins) can be reduced into much smaller single-chain Fv fragments (scFvs), ca. 1/6 as molecular mass, but which often retain the antigen-binding capacity of the parent antibodies.10–14) These scFvs can be expressed from a single transcript, and thus can be fused with a variety of functional proteins, such as enzymes and fluorescent proteins, to generate “clonable labeled antibodies” with distinct properties.15–17) Q-body, a family of innovative antibody reagents particularly suitable for on-site testing was recently developed.18,19) Q-body is antibody fragments (scFvs or Fab fragments) labeled at specific sites with fluorescent dyes, and is usually prepared by in vitro translation. Q-body fluoresces only when bound to cognate antigens, and enables homogenous, noncompetitive, sensitive, and quick detection of small molecules, which is almost impossible to perform with the conventional immunoassays.
To develop advanced immunochemical assays based on functionally modified scFvs, genes encoding variable regions should be cloned along with the immunoglobulin G (IgG)-form antibody proteins. Thus we established novel hybridoma clones producing usable antibodies against THC, amplified the genes encoding the variable regions thereof, assembled the scFv, and improved its affinity for THC via in vitro affinity maturation, i.e., “molecular breeding.”14,20)
MATERIALS AND METHODS
Cannabinoids and Their DerivativesTHC and THCA were prepared from Cannabis sativa L. in our laboratory (Kyushu University). CBN was supplied by Central Customs Laboratory. Solutions of THC-COOH and THC-COOGlu were obtained from Sigma-Aldrich. The immunogenic conjugate of THC and bovine serum albumin (THC–BSA) was purchased from GenWay Biotech.
BuffersPB: 0.050 M sodium phosphate buffer (pH 7.3); PBS: PB containing 9.0 g/L NaCl; G-PBS: PBS containing 1.0 g/L gelatin; T-PBS: PBS containing 0.050% (v/v) Tween 20; M-PBS: PBS containing 20 g/L skim milk; PVG-PBS: G-PBS containing 1.0 g/L polyvinyl alcohol with average polymerization degree 500; PBS-2: 10 mM Na2HPO4, 1.8 mM KH2PO4, 0.14 M NaCl, 2.7 mM KCl (pH 7.4); and M-PBS-2: PBS-2 containing 20 g/L skim milk.
Immunization, Cell Fusion, and Monoclonal Antibody ProductionTwo female BALB/c mice (8 weeks of age) (Japan SLC) were immunized with THC–BSA biweekly for a total of 4 times. The conjugate (25 µg) was subcutaneously injected into multiple sites on the back with a 1 : 1 emulsion (0.2 mL/mouse) of sterile saline and Freund’s complete (primary and fourth immunizations) or incomplete adjuvant (all other booster immunizations) (DIFCO). The mice then received intraperitoneal and intrasplenic injections of the conjugate (25 µg total) prepared in sterile saline, and splenocytes were collected after three days.
The cell fusion was performed according to our previous procedures.17,21) Briefly, splenocytes from two mice (4×108 cells) were fused with P3/NS1/1-Ag4-1 (NS1) myeloma cells22) (8×107 cells) using the 40% polyethylene glycol 4000 solution. Fused cells were cultured for ca. two weeks at 37°C in HAT medium supplemented with 10% Briclone (DS Pharma Biomedical) under 5% CO2/95% air. Culture supernatants were then screened by enzyme-linked immunosorbent assay (ELISA) using microplates coated with THC–BSA and blocked with M-PBS.13) To saturate co-existing anti-BSA antibodies, ELISA was performed in a PBS containing 5.0% BSA. Captured antibodies were then visualized with peroxidase (POD)-labeled goat anti-mouse IgG (Fc-specific) antibody (Jackson ImmunoResearch). Hybridomas producing anti-THC antibodies were expanded in HT medium, and then cloned by limiting dilution. Finally, the monoclonal antibodies in the culture supernatant, produced by the cloned hybridomas, were characterized. The heavy and light chain isotypes were determined with ImmunoPure Monoclonal Antibody Isotyping Kit II (ThermoFisher). Fab fragment of Ab-THC#33 was prepared with Pierce Fab Preparation Kit (ThermoFisher) for determining their antigen-binding parameters.
Cloning the Variable Region Genes of Antibody against THCThe VH and VL genes of Ab-THC#33 (IgG1κ), a monoclonal antibody established as described, were cloned as described previously.16,17) Total RNA was extracted from hybridoma #33 (ca. 1×107 cells) and reverse-transcribed using Superscript II reverse transcriptase (Invitrogen) and an oligo-dT primer. The VH fragment was amplified from the resulting cDNA with reverse primers specific for the reader sequence (#MHV-4)23) and a forward primer specific for γ1-chain (mγ1-GSP).13) This PCR was performed using Ex Taq DNA polymerase (TaKaRa Shuzo) and 35 amplification cycles [95°C (1 min), 50°C (1 min), and 72°C (1 min)] followed by an extension at 72°C (10 min). The VL gene was cloned by rapid amplification of cDNA 5′-ends (5′-RACE)24) using the cDNA as the template with a forward primer specific for κ-chain (mκ-GSP).13) The DNA fragments obtained were subcloned into pBluescript II (Toyobo) and the VH and VL nucleotide sequences therein were determined using sequencing analysis with a standard protocol.
Assembly and Expression of Wild-Type scFv GeneThe VH and VL genes of Ab-THC#33 were separately amplified from the first strand cDNAs to add a linker sequence, FLAG tag, and restriction sites.14,16) The primers used were THC#33VH-Rev and THC#33VH-For (for amplifying VH) and THC#33VL-Rev and THC#33VL-For (for amplifying VL), respectively (Table 1). Amplification products were gel-purified and spliced by overlap extension PCR13,14) to generate a gene encoding “wild-type” scFv (wt-scFv), which was then subcloned into the NcoI-SalI site in pEXmide 5,25) and propagated in Escherichia coli (E. coli) XL1-Blue cells (Stratagene).
Table 1. Nucleotide Sequences of Primers Used for PCR-Cloning of
V-Genes
| Primer | Sequence (5′→3′; Restriction sitea)) |
|---|
| THC#33VH-Rev | ATTGTTATTACTCGCGGCCCAACCGGCCATGGCCGAAGTGAAGCTGGTGGAGTCTGGG (NcoI) |
| THC#33VH-For | CCGCCGGATCCACCTCCGCCTGAACCGCCTCCACCTGAAGAGACTGTGAGAGTGGTG |
| THC#33VL-Rev | CAGGCGGAGGTGGATCCGGCGGTGGCGGATCGGACATCCAGATGACTCAGTCTC |
| THC#33VL-For | GATTTGGGCTCAACTTTCTTGTCGACTTTATCATCATCATCTTTATAATCAGCCCGTTTCAGCTCCAGCTTG (SalI) |
| THC#33VL-For-2 | GATTTGGGCTCAACTTTCTTGTCGACTTATTATTTATCATCATCATCTTTATAATC |
a) Underlined in the nucleotide sequences.
scFv mutants were generated by error-prone PCR26) as previously described.14,20) The pEXmide 5 containing the wt-scFv gene (1 ng) was mixed in a buffer solution (100 µL) with or without 0.10 mM MnCl2, THC#33VH-Rev and THC#33VH-For (or THC#33VL-Rev and THC#33VL-For) primers (Table 1) (0.10 nmol each), AmpliTaq DNA polymerase (Applied Biosystems) (5 U), and either of the following unbalanced deoxyribonucleotide triphosphates (dNTPs): 0.10 µmol of each dNTP except deoxyadenosine triphosphate (dATP) (0.020 µmol) or 0.10 µmol of each dNTP except deoxyguanosine triphosphate (dGTP) (0.020 µmol). This mixture was amplified for 35 cycles at 95°C (1 min), 50°C (1 min), and 72°C (3 min), followed by a 10 min extension at 72°C. The mutated VH and VL genes that were amplified using the same unbalanced dNTPs (the products obtained with 0 and 0.10 mM MnCl2 were combined) were spliced to produce scFv genes.13,14) The two gene libraries of the mutated scFv (“dATP-diminished VH/VL” and “dGTP-diminished VH/VL” combinations) were separately ligated into the pEXmide 5,25) and transformed into XL1-Blue cells. The resulting bacterial libraries were used to rescue phage particles, which were partially purified and examined as follows.
Selection of THC-Binding Phage ClonesNunc MaxiSorp polystyrene tubes (12×75 mm, General Laboratory Supply) were coated overnight at room temperature with 2.0 mL THC–BSA in 0.10 M carbonate buffer (pH 8.6) (5.0 µg/mL), and blocked with M-PBS-2 as described previously.13,14) In each round of panning, antigen-coated tubes were incubated with phages [ca. 1–10×1011 colony-forming unit (cfu)] in M-PBS-2 (2.0 mL) for 1 h at 37°C with continuous tumbling. Tubes were then washed three times with T-PBS-2 (4 mL), and bound phages were eluted by addition of solutions (1.0 mL) containing 10, 1.0, and 0.10 µg/mL THC in PVG-PBS for the first, second, and third round, respectively. In each round, the recovered phage suspension was added to log-phase XL1-Blue cells in 2×YT medium containing 10 µg/mL tetracycline (9 mL), and then incubated at 37°C for 30 min. After centrifugation (10000×g, 10 min), the pellet was suspended in 2×YT medium (100 µL) and spread on a 90ϕ plate containing 2×YT agar supplemented with 100 µg/mL ampicillin, 10 µg/mL tetracycline, and 10 g/L glucose, and incubated overnight at 37°C. Colonies were then scraped into 2×YT medium containing 15% glycerol (1.5 mL) and a small aliquot was used for phage rescue. The resulting phages were used in the next round of selection.
Preparation of Soluble (Non-phage-Linked) scFvsRecombinant plasmids were extracted from infected bacterial clones and scFv genes therein were PCR amplified with primers THC#33VH-Rev and THC#33VL-For-2 (Table 1) to add stop codons to their 3′-termini.14) PCR products were gel-purified, ligated into pEXmide 5, and transformed into XL1-Blue cells. Transformants were grown, and induced with isopropyl β-D-thiogalactopyranoside and sucrose.13,14) Periplasmic extracts containing soluble scFv proteins were then used for ELISA without further purification.
Affinity-Purification of scFvs and Determination of Antigen-Binding ParametersFor gel electrophoresis and determination of affinity for THC, the scFv proteins were purified by affinity chromatography using anti-FLAG-M2 agarose (Sigma-Aldrich) as described previously.14) Affinity and dissociation rate constants (ka and kd) and affinity constants (Ka) against THC residues on THC–BSA for the purified scFvs, as well as for the Fab fragment of the antibody Ab-THC#33, were determined at 25°C using BLItz (Fortebio), a biolayer interferometry sensor. Streptavidin-coated biosensor tips, which had been saturated with a biotin-labeled THC–BSA, prepared by the reaction with EZ-Link NHS-LC-Biotin (Thermo Fisher Scientific), were dipped into 4 µL solutions of scFv or Fab in PVG-PBS (10, 20, 50, or 100 nM). Association of scFvs or Fab to the THC residues was monitored for 300 s, and then dissociation was measured for 300 s in PVG-PBS.
Competitive ELISACharacterization of Mouse Anti-THC AntibodiesNinety six-well microplates (#3590, Costar) were coated overnight at room temperature with THC–BSA in 0.10 M carbonate buffer (pH 8.6) (0.50 µg/mL) and blocked with M-PBS at 37°C for 2.0 h.14) Wells were then washed three times with T-PBS, and incubated at 37°C for 1.0 h with a mixture of THC or related compounds dissolved in 50% (v/v) ethanol (25.0 µL/well) and monoclonal antibody (the hybridoma supernatants) diluted with G-PBS (100 µL/well). Subsequently, wells were washed and probed with POD-labeled goat anti-mouse IgG (Fc-specific) antibody (Jackson ImmunoResearch) diluted in G-PBS (160 ng/mL) (100 µL/well). After incubation at 37°C for 30 min, wells were washed and captured POD activity was determined colorimetrically at 490 nm using o-phenylenediamine as hydrogen donor.13,21)
Characterization of Anti-THC scFvsThe 96-well microplates were coated with THC–BSA and blocked with Block Ace (Dainippon Pharmaceutical) at 37°C for 1.0 h, and incubated at 37°C for 1.0 h with a mixture of THC or related compounds dissolved in PVG-PBS (50.0 µL/well) and soluble scFv protein diluted with PVG-PBS (100 µL/well). Wells were then washed, and probed at 37°C for 30 min with POD-labeled anti-FLAG M2 antibody (Sigma-Aldrich) diluted in PVG-PBS (0.50 µg/mL) (100 µL/well). After washing the wells, the bound POD activity was determined similarly.13,21)
In both assays described above, concentrations of antibodies and scFvs were adjusted to give bound enzyme activities at B0 of ca. 1.0–1.5 absorbance after a 30-min enzyme reaction.
RESULTS AND DISCUSSION
Production and Characterization of Mouse Monoclonal Anti-THC AntibodiesAlthough antisera against THC were generated in 1970 s27) and continue to be used,28) only few publications have so far described the production of monoclonal antibodies against THC.29–31) Thus, we here newly established antibody-secreting hybridoma clones to obtain a genetic resource that enables generation of various recombinant anti-THC immunochemical reagents. BALB/c mice, the most common donor used to generate splenocytes for establishing hybridomas, were immunized with a commercially available THC–BSA conjugate. Splenocytes from these mice were then fused with NS1 myeloma cells.22,32) A single cell fusion experiment produced four hybridoma clones #12, #15, #30, and #33, each secreting an antibody that binds to THC. These antibodies were designated Ab-THC#12 (γ1, κ), #15 (γ1, κ), #30 (γ2b, κ), and #33 (γ1, κ) with heavy and light chain isotypes denoted in the parentheses.
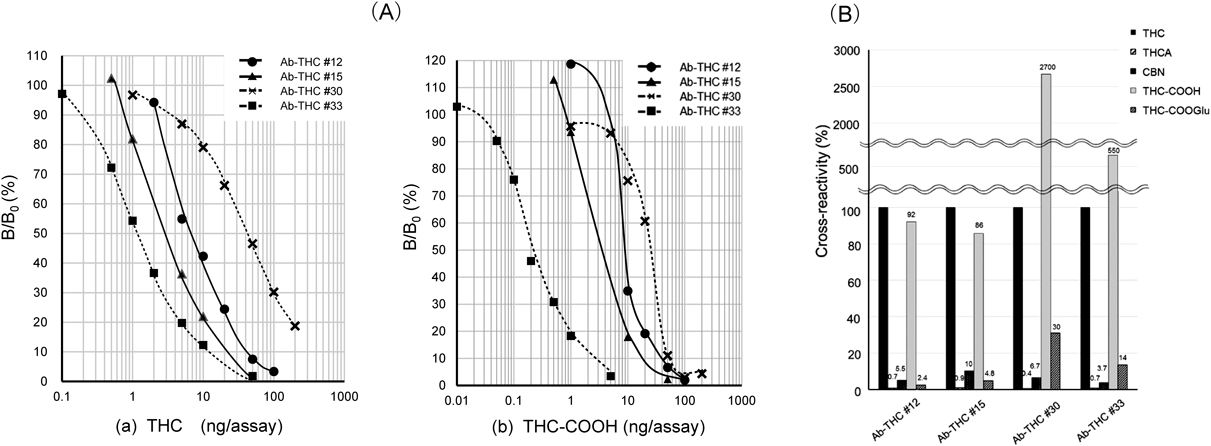
In competitive ELISA, these antibodies generated dose–response curves with different measurable ranges for THC (Fig. 2A(a)). The midpoint, which free THC inhibits antibody binding to immobilized THC by 50% and is a measure of assay sensitivity, was 7.0, 3.0, 40, and 1.1 ng/assay for Ab-THC#12, #15, #30, and #33, respectively. It should be noted that, in this study, the unit “X g/assay,” which means that totally X g (mass) of analyte was added to the assay chamber (microwells, in this case) for the competitive antigen-antibody reactions, is used to express doses of analyte or cross-reactive analogs.13–17,20,21) Thus, Ab-THC#33 exhibited the highest sensitivity, and accordingly, the highest estimated affinity for THC. Ka of the Fab fragment prepared from Ab-THC#33 was determined to be 6.2×107 M−1.
Cross-reactivity with related compounds is shown in Fig. 2B. All antibodies discriminated against THCA (<1%) and CBN (3.7–10%), while significant cross-reactivity was observed for THC-COOH (86−2700%). Cross-reactivity for THC-COOGlu varied largely among these antibodies (2.4–30%). The results for THC-COOH strongly suggest that, in the THC–BSA conjugate that we used in the immunization, BSA is probably linked to a functional group introduced at the 11-position (Fig. 1A). Unfortunately, the chemical structure of the conjugate is not publicly available. Notably, the apparent group-specificity to THC and THC-COOH may prove advantageous in practical terms, because the antibodies might not only be used to detect the marker compound of cannabinoids, i.e., THC, in materials, but may also be used to quantify THC and/or its major metabolites THC-COOH in urine.5,6) Dose–response curves constructed with THC-COOH covered ca. 40–4000 pg/assay, indicated that Ab-THC#33 enables picogram-range determination of this metabolite (Fig. 2A(b)).
Generation of Anti-THC scFvThe Substance Abuse and Mental Health Services (SAMHSA) recommends a cutoff of 50 ng/mL for detecting cannabinoids.33) Thus, the ELISA sensitivity of the monoclonal antibodies obtained should be improved, even that of Ab-THC#33. As the sensitivity of competitive immunoassays is significantly influenced by the affinity of the antibody for the antigen,14,20) we transformed Ab-THC#33 into an scFv form, suitable for genetic manipulation, and then attempted to improve its affinity for THC by introducing random point mutations. Thus, gene fragments encoding the VH and VL domains of Ab-THC#33 were cloned, and then spliced via a linker sequence encoding a (GlyGlyGlyGlySer)3 peptide. A FLAG tag34) was also added to the 3′-end to facilitate purification and detection. The resulting wt-scFv gene, with structure 5′-VH-linker-VL-FLAG, was expressed in E. coli cells. Characterization of the scFv protein obtained is described later.
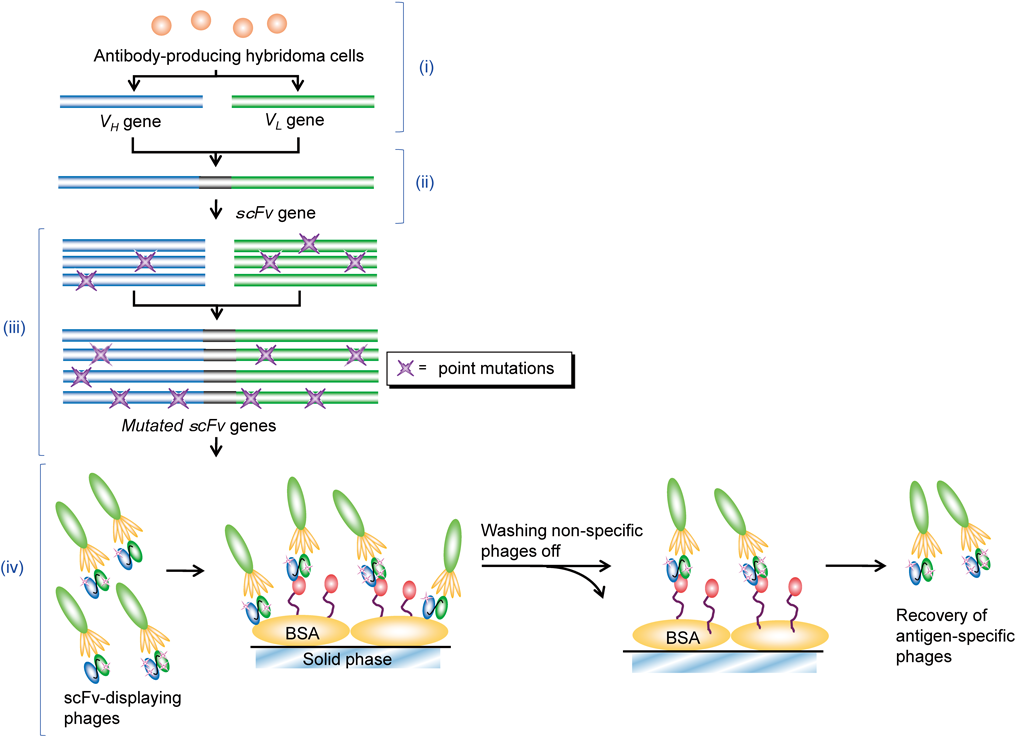
In Vitro Affinity MaturationWe previously generated an scFv mutant specific to estradiol-17β with over 150-fold higher affinity than wild-type, as measured by Ka. The mutant was generated after serial three-step mutagenesis and selection14,20) based on phage display technology,8,35) in which random point mutations in VH and VL were introduced at each step by error-prone PCR.26) This previous study demonstrated that a few mutations, not only in complementarity-determining regions (CDRs) but also in framework regions (FRs), may sometimes dramatically enhance affinity. Thus, we performed simple random mutagenesis throughout the variable regions to generate anti-THC scFv mutants with improved affinities.
The experimental flow of in vitro affinity maturation is summarized in Fig. 3. The VH and VL regions in wt-scFv gene were amplified in a single error-prone PCR under “dATP-diminished” or “dGTP-diminished” condition,20) and the resulting mutated VH and VL genes from the same condition were spliced combinatorially to create a diverse set of scFv genes. Transformation of E. coli cells with these scFv mutants generated a “dATP-diminished” and “dGTP-diminished” bacterial libraries (each containing ca. 1×106 transformants with scFv-genes). From these libraries, phage particles were separately rescued and subjected to three rounds of panning against polystyrene tubes coated with THC–BSA. Antigen-coated tubes were incubated at 37°C for 1 h with 1×1012 (the first round) or 1×1011 (the second and third round) cfu of phages to capture scFv clones with reasonable association rate constants (ka). Unbound and weakly bound phage particles were thoroughly washed out, and bound phage particles were recovered by incubating with free THC. The amount of free THC used for the recovery was progressively lowered from 10 to 1.0 µg, and then to 0.10 µg to enrich for high-affinity clones. A total of 1.1×108, 1.2×107, and 1.5×107 cfu were recovered at the first, second, and third round, respectively. After the third panning, 72 and 88 phage clones from the “dATP-diminished” and “dGTP-diminished” libraries, respectively, were screened, and consequently, a phage clone displaying an scFv mutant, named scFv#m1-36, with ca. 3-fold higher sensitivity than wild-type in ELISA assays (described later) was isolated from the latter library.
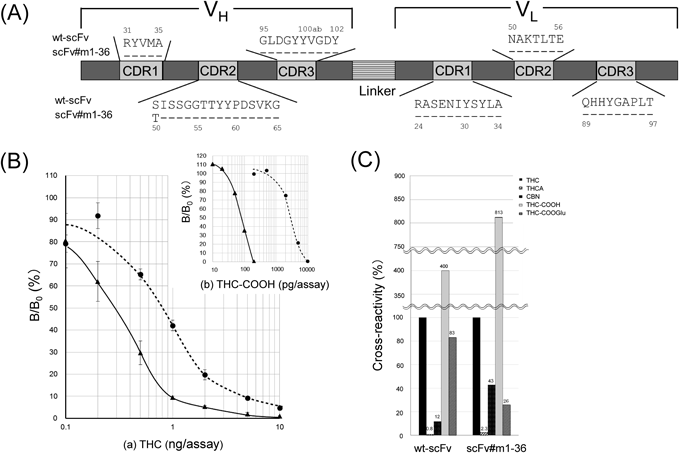
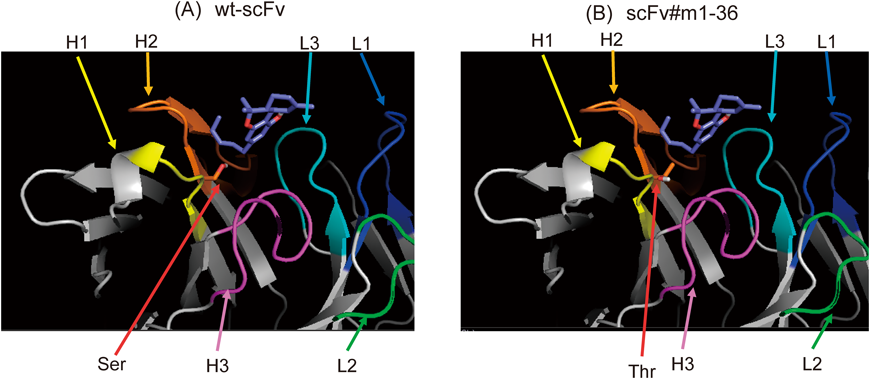
Characterization of Wild-Type and Improved scFvsThe primary structures of wild-type and mutant scFvs are illustrated in Fig. 4A, with sequences of the CDRs.36) We emphasize that a single and conservative substitution from serine (Ser) to threonine (Thr) at the N-terminus of VH-CDR2, i.e., the H50 position,36) enhanced the affinity for THC. This substitution introduces a methyl group into the paratope (antigen-binding site) of the wt-scFv, which might increase hydrophobicity and thereby enhance interaction with a hydrophobic molecule such as THC. In particular, protein modeling (Fig. 5) suggested that the introduced methyl group might be in a position to contact the pentyl side chain on THC molecule. Modeling also suggested that four CDR loops, namely VH-CDR1, VH-CDR2, VH-CDR3, and VL-CDR3 might play an important role in the interaction between THC and both wild-type and mutant scFvs.
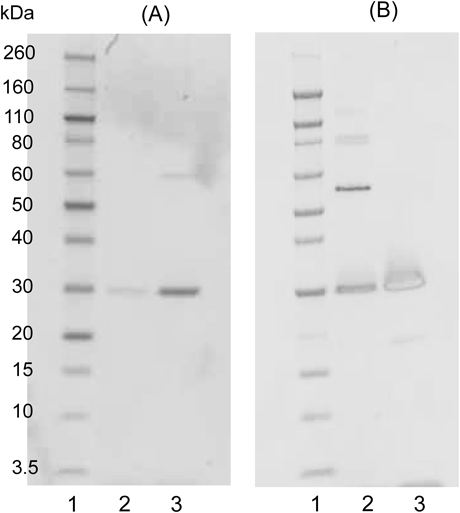
scFvs were then prepared as the soluble proteins and characterized. On sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) gels, affinity-purified wt-scFv and scFv#m1-36 proteins migrated as a single band with nearly the expected relative molecular mass (Mr) of 26805 and 26819, respectively (Fig. 6A). The immunoblotting experiments probed with anti-FLAG antibody indicated that these proteins were expressed in-frame to the end of the C-terminus (Fig. 6B).
The affinity constant (Ka) of mutant scFv for THC was 1.1×108 M−1, thus 10-fold higher than that of wild-type scFv (1.1×107 M−1). Kinetic analysis revealed that a lower dissociation rate constant (kd), 3.2×10−4 s−1 for the mutant scFv#m1-36 (2.2×10−3 s−1 for wt-scFv) mainly contributes to the enhanced affinity. The association rate constants (ka) for mutant scFv and wt-scFv were 3.6×104 M−1 s−1 and 2.3×104 M−1 s−1, respectively.
In competitive ELISA, the midpoint of the dose–response curve for THC was lower for wt-scFv (0.80 ng/assay) (Fig. 4B(a)) than for the parent mouse antibody Ab-THC#33 (1.1 ng/assay; see Fig. 2A(a)), in spite that Ka of wt-scFv was more than 5-fold lower than that of the parent antibody. The increased sensitivity with wt-scFv may be due to its monovalent structure, which is less susceptible to avidity effects that are probably present in ELISA systems on microplates with multiply immobilized antigens. Mutant scFv generated a dose–response curve with even higher sensitivity (Fig. 4B(a)): this improvement should be due to enhanced affinity. The midpoint of which was 0.27 ng/assay and was 3-fold less than that of wt-scFv (see above). The limit of detection (LOD), which was determined as the amount of THC required to give a bound absorbance that is two standard deviations below the average (n=10) of the B0 absorbance, was 0.10 ng/assay for mutant scFv, and was 2-fold less than that of wt-scFv (0.20 ng/assay).
The wild-type scFv exhibited increased cross-reactivity with CBN (3-fold) and THC-COOGlu (5.9-fold) compared with the parent antibody Ab-THC#33 (Fig. 4C). The mutant scFv showed further higher cross-reactivity for CBN (>10-fold of the parent antibody) along with moderate but significant increase in that for THC-COOH (ca. 1.5-fold) and THC-COOGlu (ca. 1.9-fold). Because CBN, THC, and cannabidiol (CBD) are the three main constituents isolated from Cannabis sativa plants37) (Fig. 1A), this increased cross-reactivity with CBN does not diminish the potential use of mutant scFv as a test for illicit cannabis-derived products.
As noted, THC is rapidly metabolized in humans to THC-COOH, which is then conjugated to THC-COOGlu. Thus, these polar metabolites are also markers of cannabis consumption.5,6) In particular, mutant scFv generated dose–response curves for THC-COOH with a picogram-order dynamic range, the midpoint of which was 80 pg/assay (Fig. 4B(b)). Therefore, mutant scFv is potentially useful not only for detecting THC, but also for monitoring the metabolite in urine at a cutoff lower than the recommended 50 ng/mL. For instance, 50 ng/mL of THC should generate ca. 50% of the B/B0 value, which would lie in the middle of the measurable range, even when specimens are diluted 10-fold to minimize matrix effects; 50 ng/mL of THC-COOH should generate the 50% B/B0 when it is assayed after ca. 30-fold dilution.
On the other hand, cross-reactivity with compounds structurally unrelated to cannabinoids, including p-acetamidophenol, acetylsalicylic acid, creatine, creatinine, caffeine, and uric acid, was negligible for both wt-scFv (<0.02%) and mutated scFv (<0.004%).
CONCLUSION
We have generated an scFv targeting THC by genetically manipulating the VH and VL genes from a newly established hybridoma that produces a mouse antibody against THC. To achieve higher sensitivity in assay systems, this wild-type scFv was then engineered by in vitro affinity maturation into a mutant scFv#m1-36, which has improved affinity in the nanomolar range, with Kd, 8.8×10−9 M. This mutant scFv will be useful to detect trace of THC and its polar metabolite, in illicit cannabis-derived materials and/or in cannabis users.
We note that based on our experience, it is challenging to obtain high-affinity antibodies against small hydrophobic molecules with few polar functional groups, such as THC. We anticipate that antibody engineering, as we have demonstrated, will overcome such difficulty in the conventional antibody productions, and promote advance in immunochemical testing for trace amounts of various molecules.
Acknowledgments
We thank Central Customs Laboratory (Kashiwa, Japan) for providing us CBN. This work was supported in part by Ushio Inc. (Chiyoda-ku, Tokyo, Japan).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Thomas BF, ElSohly MA. The Analytical Chemistry of Cannabis, 1st Edition, Elsevier, the Netherlands (2016).
- 2) Mehmedic Z, Chandra S, Slade D, Denham H, Foster S, Patel AS, Ross SA, Khan IA, ElSohly MA. Potency trends of Δ9-THC and other cannabinoids in confiscated cannabis preparations from 1993 to 2008. J. Forensic Sci., 55, 1209–1217 (2010).
- 3) Elsohly MA, Slade D. Chemical constituents of marijuana: The complex mixture of natural cannabinoids. Life Sci., 78, 539–548 (2005).
- 4) Wohlfarth A, Mahler H, Auwärter V. Rapid isolation procedure for Δ9-tetrahydrocannabinolic acid A (THCA) from Cannabis sativa using two flash chromatography systems. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci., 879, 3059–3064 (2011).
- 5) Sharma P, Murthy P, Bharath MMS. Chemistry, metabolism, and toxicology of Cannabis: clinical implications. Iran. J. Psychiatry, 7, 149–156 (2012).
- 6) Ujváry I, Grotenherman F. 11-Nor-9-carboxy-Δ9-tetrahydrocannabinol—A ubiquitous yet underresearched cannabinoid. A review of the literature. Cannabinoids, 9, 1–8 (2014).
- 7) Wong R, Tse H. Lateral Flow Immunoassay, Humana Press, New York, (2009).
- 8) Handbook of therapeutic antibodies. (Dübel S ed.) Wiley-Blackwell, Hoboken, New Jersey (2010).
- 9) Bradbury ARM, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat. Biotechnol., 29, 245–254 (2011).
- 10) Skerra A, Plückthun A. Assembly of a functional immunoglobulin Fv fragment in Escherichia coli. Science, 240, 1038–1041 (1988).
- 11) Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, Lee T, Pope SH, Riordan GS, Whitlow M. Single-chain antigen-binding proteins. Science, 242, 423–426 (1988).
- 12) Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NBM, Hamid M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol., 2012, 980250 (2012).
- 13) Kobayashi N, Shibahara K, Ikegashira K, Shibusawa K, Goto J. Single-chain Fv fragments derived from an anti-11-deoxycortisol antibody: Affinity, specificity, and idiotype analysis. Steroids, 67, 733–742 (2002).
- 14) Kobayashi N, Oyama H, Kato Y, Goto J, Söderlind E, Borrebaeck CAK. Two-step in vitro antibody affinity maturation enables estradiol-17β assays with more than 10-fold higher sensitivity. Anal. Chem., 82, 1027–1038 (2010).
- 15) Kobayashi N, Iwakami K, Kotoshiba S, Niwa T, Kato Y, Mano N, Goto J. Immunoenzymometric assay for a small molecule, 11-deoxycortisol, with attomole-range sensitivity employing an scFv-enzyme fusion protein and anti-idiotype antibodies. Anal. Chem., 78, 2244–2253 (2006).
- 16) Oyama H, Tanaka E, Kawanaka T, Morita I, Niwa T, Kobayashi N. Anti-idiotype scFv-enzyme fusion proteins: A clonable analyte-mimicking probe for standardized immunoassays targeting small biomarkers. Anal. Chem., 85, 11553–11559 (2013).
- 17) Oyama H, Morita I, Kiguchi Y, Miyake S, Moriuchi A, Akisada T, Niwa T, Kobayashi N. Gaussia luciferase as a genetic fusion partner with antibody fragments for sensitive immunoassay monitoring of clinical biomarkers. Anal. Chem., 87, 12387–12395 (2015).
- 18) Abe R, Ohashi H, Iijima I, Ihara M, Takagi H, Hohsaka T, Ueda H. “Quenchbodies”: Quench-based antibody probes that show antigen-dependent fluorescence. J. Am. Chem. Soc., 133, 17386–17394 (2011).
- 19) Abe R, Jeong HJ, Arakawa D, Dong J, Ohashi H, Kaigome R, Saiki F, Yamane K, Takagi H, Ueda H. Ultra Q-bodies: quench-based antibody probes that utilize dye–dye interactions with enhanced antigen-dependent fluorescence. Sci. Rep, 4, 4640 (2014).
- 20) Oyama H, Yamaguchi S, Nakata S, Niwa T, Kobayashi N. “Breeding” diagnostic antibodies for higher assay performance: A 250-fold affinity-matured antibody mutant targeting a small biomarker. Anal. Chem., 85, 4930–4937 (2013).
- 21) Kobayashi N, Banzono E, Shimoda Y, Oyama H, Kunihiro T, Morita I, Ohta M. A monoclonal antibody-based enzyme-linked immunosorbent assay for human urinary cotinine to monitor tobacco smoke exposure. Anal. Methods, 3, 1995–2002 (2011).
- 22) Köhler G, Howe SC, Milstein C. Fusion between immunoglobulin-secreting and nonsecreting myeloma cell lines. Eur. J. Immunol., 6, 292–295 (1976).
- 23) Jones ST, Bendig MM. Rapid PCR-cloning of full-length mouse immunoglobulin variable regions. Biotechnology, 9, 88–89 (1991).
- 24) Frohman MA, Dush MK, Martin GR. Rapid production of full-length cDNAs from rare transcripts: amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. U.S.A., 85, 8998–9002 (1988).
- 25) Jirholt P, Ohlin M, Borrebaeck CAK, Söderlind E. Exploiting sequence space: shuffling in vivo formed complementarity determining regions into a master framework. Gene, 215, 471–476 (1998).
- 26) Leung DW, Chen E, Goeddel DV. A method for random mutagenesis of a defined DNA segment using a modified polymerase chain reaction. Technique, 1, 11–15 (1989).
- 27) Teale JD, Forman EJ, King LJ, Marks V. Production of antibodies to tetrahydrocannabinol as the basis for its radioimmunoassay. Nature, 249, 154–155 (1974).
- 28) Kircher V, Parlar H. Determination of Δ9-tetrahydrocannabinol from human saliva by tandem immunoaffinity chromatography-high-performance liquid chromatography. J. Chromatogr. B Biomed. Appl., 677, 245–255 (1996).
- 29) Ullman EF, Milburn G, Jelesko J, Radika K, Pirio M, Kempe T, Skold C. Anti-immune complex antibodies enhance affinity and specificity of primary antibodies. Proc. Natl. Acad. Sci. U.S.A., 90, 1184–1189 (1993).
- 30) Tanaka H, Goto Y, Shoyama T. Monoclonal antibody based enzyme immunoassay for marihuana (cannabinoid) compounds. J. Immunoassay, 17, 321–342 (1996).
- 31) Qi L, Yamamoto N, Meijler MM, Altobell LJ 3rd, Koob GF, Wirsching P, Janda KD. Δ9-Tetrahydrocannabinol immunochemical studies: haptens, monoclonal antibodies, and a convenient synthesis of radiolabeled Δ9-tetrahydrocannabinol. J. Med. Chem., 48, 7389–7399 (2005).
- 32) Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature, 256, 495–497 (1975).
- 33) The Substance Abuse and Mental Health Services (SAMHSA).: ‹http://www.samhsa.gov/workplace/drug-testing›
- 34) Knappik A, Plückthun A. An improved affinity tag based on the flag® peptide for the detection and purification of recombinant antibody fragments. Biotechniques, 17, 754–761 (1994).
- 35) Phage display. A practical approach. (Clackson T, Lowman HB eds.) Oxford University Press, Oxfold, New York (2004).
- 36) Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of Proteins of Immunological Interest, U. S. Department of Health and Human Services, National Institutes of Health, U. S. Government Printing Office: Washington, DC (1991).
- 37) Staub C. Chromatographic procedures for determination of cannabinoids in biological samples, with special attention to blood and alternative matrices like hair, saliva, sweat and meconium. J. Chromatogr. B Biomed. Sci. Appl., 733, 119–126 (1999).
- 38) Abraham GE. Solid-phase radioimmunoassay of estradiol-17β. J. Clin. Endocrinol. Metab., 29, 866–870 (1969).
- 39) Guex N, Diemand A, Peitsch MC. Protein modelling for all. Trends Biochem. Sci., 24, 364–367 (1999).
- 40) Grosdidier A, Zoete V, Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res., 39 (Suppl.), W270–W277 (2011).