Regular Articles
Lead-Induced ERK Activation Is Mediated by GluR2 Non-containing AMPA Receptor in Cortical Neurons
2017 Volume 40 Issue 3 Pages 303-309
Details
2017 Volume 40 Issue 3 Pages 303-309
Lead is a persistent environmental pollutant and exposure to high environmental levels causes various deleterious toxicities, especially to the central nervous system (CNS). The α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor that is devoid of the glutamate receptor 2 (GluR2) subunit is Ca2+-permeable, which increases the neuronal vulnerability to excitotoxicity. We have previously reported that long-term exposure of rat cortical neurons to lead acetate induces decrease of GluR2 expression. However, it is not clarified whether lead-induced GluR2 decrease is involved in neurotoxicity. Therefore, we investigated the contribution of GluR2 non-containing AMPA receptor to lead-induced neurotoxic events. Although the expression of four AMPA receptor subunits (GluR1, GluR2, GluR3, and GluR4) was decreased by lead exposure, the decrease in GluR2 expression was remarkable among four subunits. Lead-induced neuronal cell death was rescued by three glutamate receptor antagonists, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, a non-selective AMPA receptor blocker), MK-801 (N-methyl-D-aspartate (NMDA) receptor blocker), and 1-naphthyl acetyl spermine (NAS, a specific Ca2+-permeable AMPA receptor blocker). Lead exposure activated extracellular signal-regulated protein kinase (ERK) 1/2, which was significantly ameliorated by CNQX. In addition, lead exposure activated p38 mitogen-activated protein kinase (MAPK p38), and protein kinase C (PKC), which was partially ameliorated by CNQX. Our findings indicate that Ca2+-permeable AMPA receptors resulting from GluR2 decrease may be involved in lead-induced neurotoxicity.
Lead is one of the most versatile and widely used metals in the industry. Most individuals may be exposed to lead by ingesting contaminated food and water, but accidental ingestion of contaminated soil, dust, and lead-containing paints may also be an exposure source.1) Exposure to high environmental levels of lead causes various deleterious health problems including hematotoxicity, which is the main lead-induced toxicity. Approximately 40–75% of lead in plasma binds to plasma proteins such as albumin and gamma globulin, and protein non-binding lead binds to low-molecular-weight thiol-containing compounds such as cysteine or homocysteine.2) The mechanism of lead-induced hematotoxicity is known to involve inhibition of δ-aminolevulinic acid dehydratase, the enzyme involved in heme biosynthesis.3) Schwartz et al. performed a cross-sectional epidemiological study to assess the relationship between blood lead levels (BLL) and anemia in children living near a primary lead smelter in 1974. This study led them to conclude that there is a positive correlation between BLL and the incidence rate of anemia because the symptoms of anemia were not observed at blood lead levels below 20 µg/dL. However, the probability of developing anemia was 2, 18, and 40% at lead levels of 20–39, 40–60, and >60 µg/dL, respectively.4)
Lead is also known to induce neurotoxicity,5–7) which manifests as symptoms including behavioral problems, decreased cognitive activity, and lower intelligence test scores. Additionally, it was reported that increased blood lead level caused by exposure to lead during developmental stages is a risk factor for attention-deficit hyperactivity disorder (ADHD).8) Although there is no discernible threshold for the adverse effects of lead, intellectual deficits associated with lead occur at BLL <10 µg/dL.9) Considering that lead induces neurotoxicity at lower concentrations than that at which it induces hematotoxicity, it is important to clarify the mechanisms of lead-induced neurotoxicity.
Glutamate, an important excitatory neurotransmitter in the central nervous system (CNS), is required for neuronal growth, maturation, and synaptic plasticity. Ionotropic glutamate receptors are divided into N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptors. Overactivation of glutamate receptors increases cytosolic Ca2+, which disrupts Ca2+ homeostasis and finally induces cell death.10) Although NMDA receptors are highly permeable to Ca2+, AMPA receptors containing the glutamate receptor 2 (GluR2) subunit are impermeable to Ca2+ under normal conditions. The Ca2+ permeability of AMPA receptors is dependent on the GluR2 subunit and AMPA receptors not containing the GluR2 subunit are Ca2+-permeable and increase the neuronal vulnerability to excitotoxicity.11)
We previously reported that long-term exposure of rat cortical neurons to lead acetate decreases GluR2 expression.12) However, the direct relationship between lead-induced GluR2 decrease and neurotoxicity have been unclear. Thus, additional research is needed to determine whether Ca2+-permeable (GluR2 non-containing) AMPA receptors are involved in lead-induced disruption of intracellular signaling and subsequent neuronal death. Therefore, we examined the role of AMPA receptors in lead-induced neuronal death.
Eagle’s minimal essential salt medium (Eagle’s MEM) was purchased from Nissui Pharmaceutical (Tokyo, Japan). Fetal calf serum (FCS) was purchased from Nichirei Biosciences Inc. (Tokyo, Japan). Horse serum (HS) was purchased from Gibco (Life Technologies, Carlsbad, CA, U.S.A.). Trypan blue, D-(+)-glucose, sodium bicarbonate (NaHCO3), sodium dodecyl sulfate (SDS), glycerol, 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), and paraformaldehyde were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Lead acetate was purchased from EBISU (Osaka, Japan). 4-(2-Hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) was purchased from DOJINDO (Kumamoto, Japan). L-Glutamine, arabinosylcytosine, formaldehyde, 1-naphthylacetylspermine (NAS), and anti-β-actin antibody (AC-15) were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.). Pentobarbital was purchased from Kyoritsu (Tokyo, Japan). Bromophenol blue was purchased from Katayama Chemical Industries Co., Ltd. (Osaka, Japan). Tris–HCl, nonidet P-40, ethylenediaminetetraacetic acid (EDTA), mercaptoethanol, and the protease inhibitor cocktail were purchased from Nacalai Tesque (Kyoto, Japan). Anti-microtubule-associated protein 2 (MAP2, MAB3418), anti-GluR2 (MAB397), and anti-GluR1 (MAB2263) antibodies were purchased from Millipore (Billerica, MA, U.S.A.). Anti-GluR3 (#4676), anti-GluR4 (#8070), anti-phospho-p44/42 mitogen-activated protein kinase (MAPK, extracellular signal-regulated protein kinase [ERK]1/2, Thr202/Tyr204, #9101), anti-p44/42 MAPK (ERK1/2, #9102), anti-phospho-p38 MAPK (Thr180/Tyr182, #9211), p38 MAPK (#9212), anti-phospho-stress-activated protein kinases (SAPK)/c-Jun amino-terminal kinase (JNK, Thr183/Tyr185, #9251), and anti-SAPK/JNK (#9258) antibodies were purchased from Cell Signaling (Danvers, MA, U.S.A.). (+)-MK-801 hydrogen maleate was purchased from RBI-Funakoshi (Tokyo, Japan).
Cell CultureThe present study was approved by the Animal Ethics Committee of Hiroshima University. The following procedures were performed under sterile conditions. The prefrontal part of the cerebral cortex was dissected out of fetal rats at gestation day 18, and the cells were dissociated by gentle pipetting, and then plated on culture plates (4×105 cells/cm2). The cultures were incubated in Eagle’s MEM supplemented with 10% FCS, L-glutamine (2 mM), D-(+)-glucose (11 mM), NaHCO3 (24 mM), and HEPES (10 mM). The cultures were maintained at 37°C exposed to an atmosphere of 5% CO2 in MEM containing 10% FCS (days in vitro [DIV] 1–7) or 10% HS (DIV 8–11) changed every 2 d. Arabinosylcytosine (10 µM) was added after DIV 6, and the cultures were used for experiments at DIV 11. This protocol has been confirmed to produce cultures containing approximately 90% neurons by immunostaining using the neuron marker, MAP2.
Trypan Blue AssayAfter drug treatment, the neurons were stained with 1.5% trypan blue for 10 min, fixed with 10% formalin for 2 min, and then rinsed with physiological saline. The stained and unstained cells were regarded as dead and viable, respectively. Cell viability was then calculated as the percentage ratio of unstained cells to total cells counted, and >200 cells/well were randomly counted.
Western BlottingAfter lead acetate treatment, the cells were washed with phosphate-buffered saline (PBS) buffer and lysed in Tris-hydrochloride (HCl)–sodium chloride (NaCl)–EDTA (TNE) buffer containing 50 mM Tris–HCl, 1% nonidet P-40, 20 mM EDTA, protease inhibitor cocktail (1 : 100), and 1 mM sodium orthovanadate. The mixture was rotated at 4°C and centrifuged at 15000 rpm, after which the supernatant was transferred to a microtube. The supernatant was added to denaturing buffer (100 mM Tris–HCl, 4% SDS, 20% glycerol, 0.004% bromophenol blue, and 5% mercaptoethanol) at 95°C for 3 min, separated using electrophoresis, and then transferred to a polyvinylidene difluoride membrane. The membranes were subsequently blocked with 5% skim milk for 1 h and incubated with primary antibody overnight, followed by incubation with the secondary antibody for 1 h. Proteins were detected using an enhanced chemiluminescent detection system (Chemi-Lumi One L, Nacalai Tesque, Kyoto, Japan) and quantified using a digital imaging software (ImageJ, NIH, Bethesda, MD, U.S.A.).
Protein Kinase C (PKC) AssayPKC activity was measured using a PKC kinase activity kit (Enzo Life Sciences, Farmingdale, NY, U.S.A.) according to the manufacturer’s instructions. Briefly, after drug treatment, the cells were lysed in lysis buffer containing 20 mM 3-(N-morpholino)propanesulfonic acid (MOPS), 50 mM β-glycerophosphate, 50 mM sodium fluoride (NaF), 1 mM dithiothreitol (DTT), and protease inhibitor cocktail (1 : 100). The mixture was centrifuged at 13000 rpm for 15 min, and then the supernatant was transferred to a microtube. Crude protein (10 ng) was measured per assay.
ImmunocytochemistryCells were seeded on polyethyleneimine-coated coverslips, incubated overnight, treated with drugs, washed with PBS, and then fixed with 4% paraformaldehyde in PBS for 10 min. The coverslips were washed with PBS, blocked with 3% bovine serum albumin (BSA) for 1 h, and incubated with anti-phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) and anti-MAP2 antibodies diluted in PBS for 1 h at room temperature. After washing thrice with PBS, the coverslips were incubated with Alexa Fluor® 488 and Alexa Fluor® 555 (both Molecular Probes®) for 1 h at room temperature in the dark. The coverslips were further washed thrice with PBS, incubated with 4′,6-diamidino-2-phenylindole (4′-6-diamidino-2-phenylindole (DAPI), Molecular Probes®) diluted in PBS for 5 min, and then washed again thrice with PBS. Finally, the coverslips were enclosed in Prolong® Diamond (Molecular Probes®) and observed under a confocal laser scanning microscope (Carl Zeiss, LSM5 PASCAL).
StatisticsAll experiments were performed at least thrice, and representative data are shown. The data are expressed as the mean±standard error of the mean (S.E.M.). The data were statistically evaluated using an ANOVA followed by Tukey’s test. A p<0.05 was considered significant.
We previously showed that GluR2 expression in primary cortical neurons was decreased by lead exposure.12) However, it is unclear whether expression of other AMPA receptor subunits was affected by lead exposure. Therefore, we investigated the effect of lead on AMPA receptor subunit expression. Rat cortical neurons were exposed to 5 µM lead for 1, 3, 5, 7, and 9 d, and then protein expression was measured using Western blotting. Although the expression of four subunits (GluR1, GluR2, GluR3, and GluR4) was time-dependently decreased by lead exposure, the decrease in GluR2 expression was remarkable among four subunits. These results suggest that exposure to lead decreases GluR2 expression in cortical neurons with slight decrease in other subunits (Fig. 1).
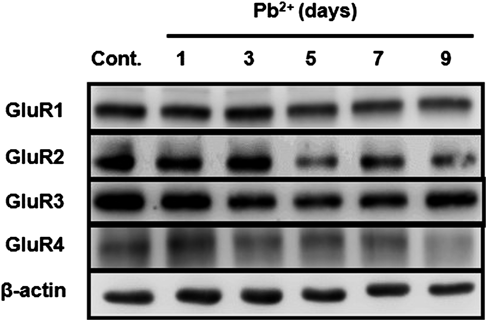
Cortical neurons were exposed to 5 µM lead acetate (Pb2+) for 1–9 d and GluR1, GluR2, GluR3, and GluR4 were detected using Western blotting.
It is unclear whether glutamate receptors are associated with lead-induced neuronal death. Therefore, to investigate the involvement of glutamate receptors in lead-induced neuronal death, cortical neurons were exposed to 5 µM lead for 9 d with or without glutamate receptor antagonists and the cell viability was evaluated. Because GluR2 decrease was observed on day 5 of exposure to lead, the cells were treated with the glutamate receptor antagonists for the last 5 d. Exposure of neurons to 5 µM lead for 9 d decreased the cell viability to approximately 60% of the control, which was attenuated by CNQX (a non-selective AMPA receptor blocker), MK-801 (an NMDA receptor blocker), and 1-naphthyl acetyl spermine (NAS, a specific Ca2+-permeable AMPA receptor blocker) (Figs. 2A–C, respectively). These results suggest that the lead-induced neuronal death was mediated by NMDA receptors and Ca2+-permeable (GluR2 non-containing) AMPA receptors.
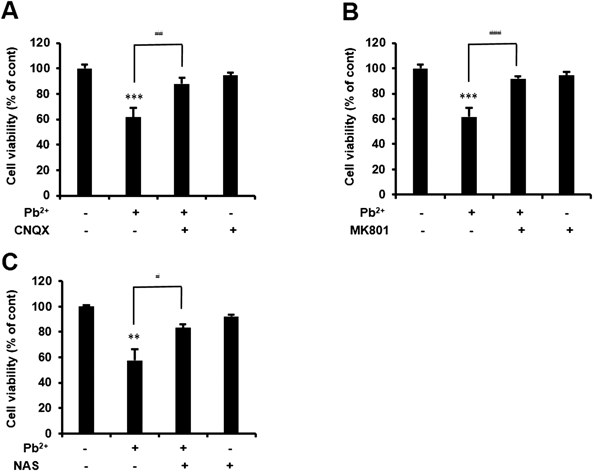
Cortical neurons were exposed to 5 µM lead acetate (Pb2+) for 9 d with or without three kinds of antagonists, (A) 20 µM CNQX, (B) 5 µM MK801, and (C) 100 µM NAS for last 5 d. Then, cell viability was measured using trypan blue assay. Data are the mean±S.E.M. (n=3–4). ** p<0.01 and *** p<0.001 vs. control. # p<0.05, ## p<0.01, and ### p<0.001.
MAPKs are a conserved family of enzymes that link extracellular signals to fundamental intracellular processes and regulate various cellular activities including growth, proliferation, differentiation, and apoptosis.13) There are three major groups of MAPKs; ERK 1/2, p38 MAPKs (p38), and JNKs. We sought to determine the signaling mechanisms underlying lead-induced neuronal cell death and the contribution of AMPA receptors to these events. Therefore, we investigated the effect of lead, CNQX, or both on the phosphorylation of ERK, p38, and JNK. Rat cortical neurons were exposed to 5 µM lead for 9 d with or without CNQX for the last 5 d, and protein expression was measured. Exposure to 5 µM lead for 9 d significantly increased phosphorylated ERK by 3.7-fold and this effect was significantly abolished by CNQX treatment (Fig. 3A). Moreover, exposure to 5 µM lead for 9 d significantly increased phosphorylated p38 by 2.9-fold and this effect was partially abolished by CNQX treatment (Fig. 3B). In contrast, phosphorylated JNK was not affected by lead exposure (Fig. 3C).
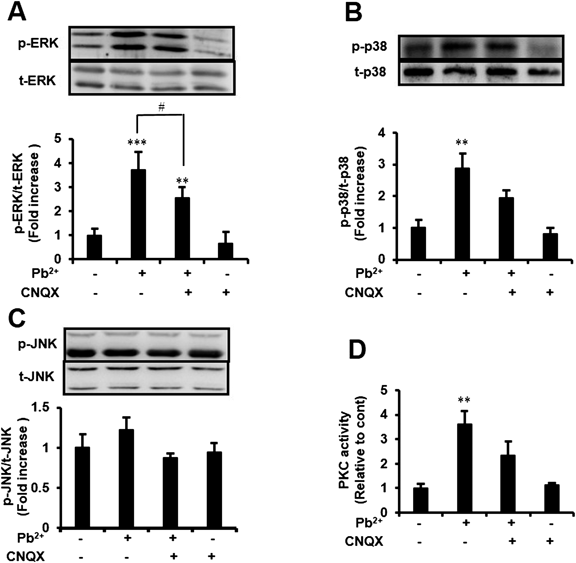
Cortical neurons were exposed to 5 µM lead acetate (Pb2+) for 9 d with or without 20 µM CNQX for last 5 d. Then, (A) phosphorylated or total ERK, (B) p38, and (C) JNK were detected using Western blotting. (D) PKC activity was measured using PKC kinase activity kit. Data are the mean±S.E.M. (n=3–4). ** p<0.01 and *** p<0.001 vs. cont. # p<0.05.
We immunostained neurons with phospho-ERK and MAP2 antibodies to confirm the inhibitory effect of CNQX on lead-induced ERK phosphorylation. As shown in Fig. 4, the activation of ERK by lead exposure was also demonstrated by an increase in the number of neurons stained positive for phospho-ERK, which was decreased by CNQX co-treatment and corroborated the results in Fig. 3A.
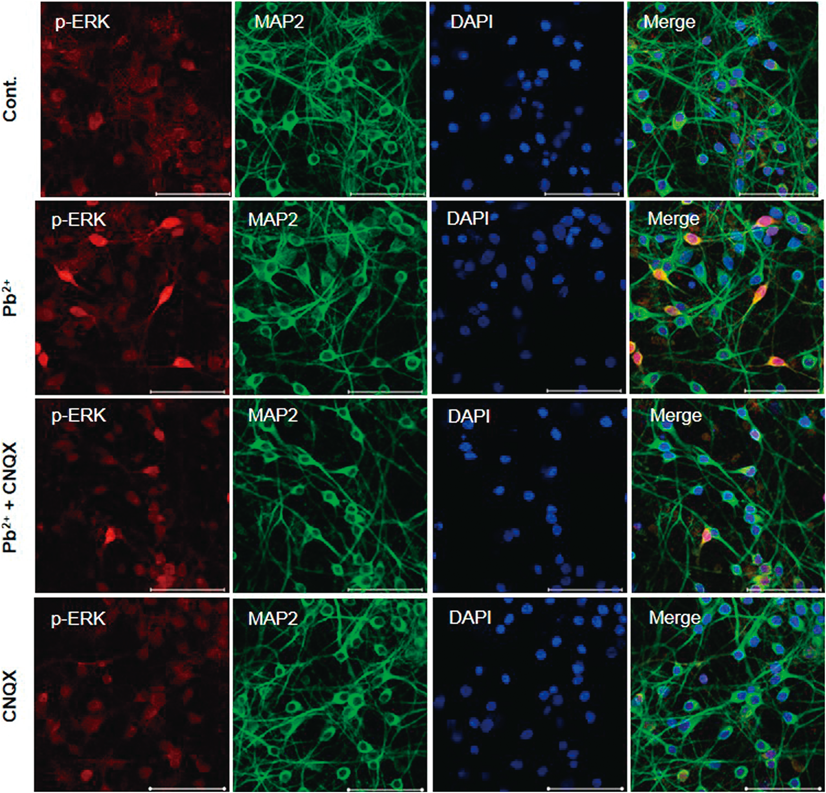
Cortical neurons were exposed to 5 µM lead acetate (Pb2+) for 9 d with or without 20 µM CNQX for last 5 d, and then immunostained with rabbit anti-phospho-p44/42 MAPK (ERK1/2), (Thr202/Tyr204) (red) and mouse anti-MAP2 (green) antibodies. 4′,6-Diamidino-2-phenylindole (DAPI) was used for nuclear staining (blue); scale bar=50 µm.
PKC, a serine/threonine protein kinase is involved in various intracellular signal pathways. Lead is known to activate PKC.14,15) Therefore, we determined the effect of lead, CNQX, or both on PKC activity. Cortical neurons were exposed to 5 µM lead for 9 d with or without CNQX for the last 5 d, and PKC activity was measured. As shown in Fig. 3D, PKC activity was significantly increased 3.6-fold by 5 µM lead exposure and this effect was partially abolished by CNQX treatment. These results suggest that ERK, p38, and PKC are activated by long-term exposure to lead, and this activation is mediated by AMPA receptors.
In the present study, we demonstrated that long-term exposure to lead activated ERK, p38, and PKC and subsequently induced neuronal cell death, which was blocked by glutamate receptor antagonists. GluR2 expression in cortical neurons was specifically decreased by lead exposure (Fig. 1). AMPA receptors are heterotetrameric complexes composed of four subunits (GluR1 to GluR4), which are encoded by separate genes. In the cortical, hippocampal pyramidal, and granule neurons most AMPA receptors are impermeable to Ca2+ because they contain the GluR2 subunit.16–19) Currently, some studies have reported that lead enters adrenal medullary cells through the L-type voltage-dependent Ca2+ channel (VDCC),20,21) which is essential for coupling membrane depolarization to the influx of Ca2+ in excitable cells. Similar to VDCCs, Ca2+-permeable AMPA receptors, resulting from decreased GluR2 expression induced by lead exposure would be permeable to lead. Interestingly, we demonstrated the protective effects of CNQX against lead-induced activation of MAPKs, PKC (Figs. 3, 4), and neuronal cell death (Fig. 2), which raise the possibility that the lead entered the neurons through AMPA receptors. Namely, it is considered that first non-toxic levels of lead influx through some uptake systems including VDCC was occurred, and then GluR2 expression was decreased, which subsequently resulted in toxic levels of lead influx through GluR2 non-containing AMPA receptors.
GluR2 knockdown animal models show neuronal vulnerability to systemic kainic acid (known as a neurotoxic glutamate analog) injection22,23) or global ischemia. In addition, the neuronal cell death observed in the GluR2 knockdown rat brain was abolished by injecting glutamate receptor antagonists, NAS and CNQX.24) Furthermore, the lead-induced decrease in cell viability was abolished by three kinds of glutamate receptor antagonists, CNQX, NAS, and MK801 (Figs. 2A–C, respectively). The activation of AMPA receptors induces membrane depolarization and relieves the blockade of NMDA channels by Mg2+, which results in Ca2+ influx through NMDA receptors.25,26) Therefore, both the increased Ca2+-permeability of AMPA receptors and subsequent activation of NMDA receptors could contribute to lead influx and its subsequent neurotoxicity.
The activation of MAPKs has been implicated in neuronal cell death induced by chemicals and heavy metals including lead. Wang et al.27) showed that exposure of the rat cortex to lead induced apoptosis through the MAPK signaling pathway by phosphorylation of ERK and JNK. In addition, Engstrom et al.28) reported that lead-induced neurotoxicity in neural precursor cells is mediated by the activation of JNK and p38 and inhibition of prosurvival Akt signaling.28) In the present study, activation of ERK (Figs. 3A, 4) and p38 (Fig. 3B) was induced by lead exposure; however, the phosphorylated JNK levels were not changed (Fig. 3C), probably because JNK activation was recovered by day 9 of lead exposure. Lead is a strong activator of PKC,14,15) and the overactivation of PKC is also known to be one of the mechanisms of lead-induced cytotoxicity. Lead affects a number of basic neuronal developmental and cognitive functions through PKC or its substrates29,30) and the activation of PKC by long-term exposure to lead was also shown in Fig. 3D.
In addition to lead, we reported that some environmental pollutants increase neuronal vulnerability by decreasing GluR2 expression.31–33) Long-term exposure to endogenous levels of tributyltin (TBT) decreases GluR2 expression in primary cortical neurons.34) Furthermore, glutamate-induced Ca2+ influx increased in TBT-treated neurons compared to that in control neurons, which increased neuronal susceptibility to glutamate excitotoxicity.34) Moreover, perfluorooctane sulfonate (PFOS) decreases GluR2 expression and enhances neuronal vulnerability to excitotoxicity in vivo and in vitro. GluR2 expression in rat cerebral cortex was decreased by developmental exposure to PFOS, and neuronal death was induced by kainate (a glutamate analog) injection into PFOS-exposed rat.35) These lines of evidence suggest that exposure to a part of chemicals affects the components of AMPA receptor subunit by decreasing GluR2. This effect increases the Ca2+-permeability of AMPA receptors, which finally induces neuronal vulnerability to excitotoxicity in the mammalian brain. In summary, long-term exposure to lead activates ERK, p38, and PKC and subsequently induces neuronal cell death, which may be caused by the lead that enters the Ca2+-permeable AMPA receptors. To date, there are no useful probes available for monitoring intracellular lead concentrations. Therefore, to confirm the direct entry of lead through GluR2 non-containing AMPA receptors, intracellular lead concentrations should be measured by inductively coupled plasma mass spectrometry (ICP-MS) in further studies. The lack of GluR2 expression has been reported to impair novelty-induced exploratory activities, motor coordination, and cognitive functions in mice.36,37) Therefore, neurotoxicity, which is characterized by symptoms such as behavioral disorders, decreased cognitive activity, and lower than normal intelligence test scores observed in patients with lead poisoning can at least be partially explained by a decrease in GluR2 expression.
This study was supported by JSPS KAKENHI (B) Grant Numbers 23310047 (to Y.K.), 15H02826 (to Y.K.), and a Grant-in-Aid for JSPS Fellows Number 14J06534 (to K.I.).
The authors declare no conflict of interest.