Abstract
Silibinin is the main constituent of silymarin, an extract from the seeds of milk thistle (Silybum marianum). Because silibinin has many pharmacological activities, extending its clinical use in the treatment of a wider variety of diseases would be desirable. In this study, we report on the binding of silibinin to plasma proteins, an issue that has not previously been extensively studied. The findings indicated that silibinin mainly binds to human serum albumin (HSA). Mutual displacement experiments using ligands that primarily bind to sites I and II clearly revealed that silibinin binds tightly and selectively to site I (subsites Ia and/or Ic) of HSA, which is located in subdomain IIA. Thermodynamic analyses suggested that hydrogen bonding and van der Waals interactions are major contributors to silibinin–HSA interactions. Furthermore, the binding of silibinin to HSA was found to be decreased with increasing ionic strength and detergent concentration of the media, suggesting that electrostatic and hydrophobic interactions are involved in the binding. Trp214 and Arg218 were identified as being involved in the binding of silibinin to site I, based on binding experiments using chemically modified- and mutant-HSAs. In conclusion, the available evidence indicates that silibinin binds to the region close to Trp214 and Arg218 in site I of HSA with assistance by multiple forces and can displace site I drugs (e.g., warfarin or iodipamide), but not site II drugs (e.g., ibuprofen).
Silymarin is an extract from the seeds of the milk thistle (Silybum marianum), and is mainly composed of flavonolignans, including silibinin (Fig. 1), isosilybin, silychristin and silidianin.1–3) Because of its hepatoprotective effects, it has been used for centuries for treating liver disorders such as hepatitis, cirrhosis, alcoholic liver disease and amatoxin mushroom poisoning.1,3,4) Silymarin and its major constituent, silibinin, are considered to be safe because there are only few reports on adverse effects.4–6) Therefore, this extract is widely consumed as a dietary supplement for liver protection world-wide.1,4,6,7) The mechanisms responsible for these actions of silymarin and silibinin are known to involve the several events, including enhanced protein synthesis through the stimulation of polymerase I and ribosomal RNA transcription, protecting the cell membrane from radical-induced damage by antioxidant activity, and the blockage of the uptake of toxins by inhibiting their binding to hepatocytes.1,3,6) Furthermore, their anti-inflammatory and antifibrotic activities are also thought to contribute to their hepatoprotective effects.3,5)
Silymarin or silibinin has recently been reported to function as anticancer agents.6–9) There are numerous studies suggesting the broad-spectrum efficacy of these compounds in inhibiting cancer metastasis.6–9) The anticancer effects of these compounds are thought to involve cell cycle arrest at the G1/S-phase, the induction of cyclin-dependent kinase inhibitors, the down-regulation of anti-apoptotic gene products, the inhibition of cell-survival kinases, and the inhibition of inflammatory transcription factors.8) Silymarin and silibinin also down-regulate gene products that are associated with the proliferation of tumor cells, invasion, angiogenesis and metastasis.7,8) In addition, the preventive activities of silymarin and silibinin against gastrointestinal problems, nephropathy, cardio-plumonary problem have also been suggested.5) Taking the multiple pharmacological effects of silibinin into consideration, it is likely that it will be widely used in treating a wide variety of diseases not only as a dietary supplement but as a prescribed drug.
Human serum albumin (HSA) is a monomeric protein consisting of 585 amino acid residues, with a molecular weight (MW) of approximately 66500 Da.10) HSA is a major protein component of blood plasma and functions as a carrier for numerous endogenous and exogenous compounds.11) HSA contains two distinct ligand binding sites, which are referred to as sites I and II.12,13) X-Ray crystallographic analyses indicate that sites I and II are located in subdomains IIA and IIIA of HSA, respectively.14–16) The distribution and pharmacological actions of silibinin are predicted to be controlled by their binding to HSA in the systemic circulation, if HSA is the major binding protein for silibinin in human plasma. Therefore, clarification of the binding protein, binding site and binding mode of silibinin are essential in predicting its pharmacokinetics and therapeutic effects and its potential interactions with other pharmaceutical agents. Maiti et al. investigated the binding of silibinin to HSA using spectroscopic techniques aided by docking studies, and proposed that the silibinin molecule lies within hydrogen bonding distance of the Trp214 and Asp415 residues of subdomains IIA and IIIA.17) However, the issue of whether HSA is the major binding protein in plasma or whether silibinin interacts with drugs that bind to sites I and II remains unclear. Thus, in spite of the fact that silibinin is in widespread use and will likely be extended to variety of diseases, it is surprising that the state of our knowledge regarding its binding to plasma proteins is limited.
In present study, we report on a systematic study of the protein binding of silibinin. HSA was first identified as the main protein in human plasma that binds silibinin. We also determined the binding parameters for silibinin to HSA. The effects of temperature, ionic strength and detergent on silibinin–HSA interactions were also investigated. Based on these findings, we propose that multiple types of interactions contribute to silibinin–HSA binding. In addition, the binding site and the amino acid residues involved in silibinin binding were also identified using displacement of site marker ligands in conjunction with the binding of silibinin to chemically modified- and mutant–HSAs. We conclude that silibinin binds to the region close to Trp214 and Arg218 in site I and mainly interacts with sites I drugs, but not site II drugs.
MATERIALS AND METHODS
MaterialsSilibinin, HSA (fraction V, fatty acid free), α1-acid glycoprotein (AAG), γ-globulins, warfarin, iodipamide, dansyl-L-asparagine (DNSA) and 2-hydroxy-5-nitrobenzyl bromide (HNB) were purchased from Sigma Chemical Co. (St. Louis, MO, U.S.A.). Sodium chloride (NaCl) and polyoxyethylene lauryl ether (PLE, hydroxyl number; 40–60) were obtained from Nacalai Tesque, Inc. (Kyoto, Japan) and Cosmo Bio Co., Ltd. (Tokyo, Japan), respectively. Ibuprofen was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Human plasma was obtained from Interstate Blood Bank, Inc. (Memphis, TN, U.S.A.). All other chemicals from commercial sources were of the highest grade. Sixty-seven micromole sodium phosphate buffer (pH 7.4) was used in the protein binding experiments.
Determination of Protein Binding of SilibininThe binding of silibinin to proteins was examined using ultrafiltration and equilibrium dialysis techniques. Ultrafiltration was carried out using Amicon® Ultra-0.5 mL centrifugal filter unit with an Ultracel®-30 membrane (Merck Millipore Co., MA, U.S.A). Samples of 500 µL were centrifuged at 2500×g at 25°C for 5 min. This procedure was repeated 5 times in order to minimize adsorption to the membrane. The samples that remained and the filtrates were discarded until the fourth centrifugation, and the concentration of free (unbound) ligand in the filtrates was quantified by HPLC after the fifth centrifugation step. Equilibrium dialysis experiments were performed using 2 mL Sanko plastic dialysis cells (Fukuoka, Japan). The two cell compartments were separated by Visking cellulose membranes. Aliquots (1.5 mL) of samples were dialyzed for 12 h against the same volume of buffer solution. After reaching equilibrium, the ligand concentrations in the buffer compartment (free ligand concentration) and in the protein compartment (free+bound ligand concentration) were determined by HPLC. The volume shift after equilibrium dialysis was corrected according to the method of Giacomini et al.18)
Quantitative Analysis of Binding DataData regarding protein binding were analyzed quantitatively. The free (unbound) fractions were calculated using ultrafiltration data, as follows.
 | (1) |
where
Cf is the free ligand concentration determined by measurement of the filtrate.
Cb is the bound ligand concentration and was calculated by subtracting
Cf from the total ligand concentration (before ultrafiltration) (
Ct). For equilibrium dialysis,
Cb was calculated by subtracting the ligand concentration in the buffer compartment (
Cf) from the ligand concentration in the protein compartment (
Cf+
Cb). Binding parameters of ligands to HSA were obtained with the data from ultrafiltration and equilibrium dialysis. The experimental data were fitted to the following equation using GraphPad PRISM
® Version 7 (GraphPad Software, Inc., CA, U.S.A.).
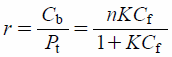 | (2) |
where
r is the number of moles of ligand bound per mole of protein.
Pt is the protein concentration. Binding parameters,
n and
K are the number of binding sites and the association constant for the high-affinity binding site, respectively. All experiments and analyses were performed using the condition,
r<0.4, to minimize ligand binding to any low-affinity binding sites.
Interaction Mode of the Two Ligands at High-Affinity Binding Sites on HSAIn order to simultaneously estimate the interaction mode between two ligands, A and B, which are binding to each primary binding site of HSA, the data were treated according to the method of Kragh–Hansen.19,20) In this method, the number of moles of ligands A and B bound per mole of protein, rA and rB can be described as follows:
 | (3) |
 | (4) |
where
Ab and
Bb are the concentration of bound ligands
A and
B, respectively.
KA and
KB are the binding constants of ligand
A and
B, respectively.
Af and
Bf are the free concentration of ligands
A and
B, respectively. χ is the coupling constant. In these equations, the independent binding of the two ligands is characterized by χ=1, while a competitive interaction results in χ=0. χ>1 and 0<χ<1 express cooperative- and anti-cooperative interaction between ligands
A and
B on the protein, respectively.
Thermodynamic Analysis of Silibinin Binding to HSAThermodynamic parameters were calculated by the method of Pederson et al.21) using van’t Hoff plots taken at four specified temperatures, 18, 25, 30 and 35°C. From the temperature dependence of the association constants obtained in the equilibrium dialysis experiments, it is possible to calculate the values for the thermodynamic parameters involved in the binding process. If the enthalpy change, ΔH does not vary significantly over the temperature range studied, ΔH and the entropy change, ΔS can be both determined by the van’t Hoff equation below,
 | (5) |
where
K is the association constant at temperature
T (absolute temperature) and
R is the gas constant. Δ
H and Δ
S were obtained by plotting the association constants determined using the equation, and the free energy change, Δ
G was calculated from the following relation.
 | (6) |
The HPLC system used in this study consisted of a Hitachi model D-2000 Elite HPLC system (Hitachi Co., Tokyo, Japan). YMC-PACK ODS AM-303 (5 µm particle size, 250×4.6 mm i.d., YMC Co., Kyoto, Japan) was used as the stationary phase and was maintained at 40°C. Two solvents, solvent A (50 mM sodium dihydrogen phosphate) and solvent B (50 mM sodium dihydrogen phosphate and acetonitrile (30 : 70, v/v)) were used as the mobile phases. The following linear gradient elution of the solvents was programmed for the quantitation of silibinin, warfarin, ibuprofen, iodipamide and DNSA: 0–7 min (30–100% B), 7–10 min (100% B), 10–15 min (100–30% B). The flow rate of the mobile phase was maintained constant at 1.0 mL/min. The detection wavelength was fixed at 210 nm and the effluent was monitored during 15 min for each sample.
Modification of AlbuminThe lone tryptophan residue, Trp214, of HSA was modified according to the method of Fehske et al.22) In a typical experiment, 1 g of HSA was dissolved in 200 mL of a 10 M urea solution and the pH was adjusted to 4.4 by adding acetic acid. HNB in 25 mL methanol was then added and the resulting solution was subjected to occasional shaking. After 2 h, the supernatant was dialyzed against water for 60 h, and the resulting solution was then lyophilized. The modification ratio, determined spectrophotometrically,22) indicated that 93% of the tryptophan residues had been modified, indicating that Trp214 in HSA was essentially completely modified. The selective modification of tyrosine residue, Tyr411, of HSA was carried out according to the method of Hagag et al.23) In a typical run, 375 µL of 20 mM p-nitrophenyl anthranilate in acetonitrile was added under continuous stirring to 60 mL of 100 µM HSA in 0.1 M sodium phosphate buffer (pH 8.0). The reaction was allowed to proceed for 7 h and the preparation was dialyzed against water for 60 h, and then lyophilized. It is known that, in this modified procedure, a single anthraniloyl moiety is selectively incorporated into Tyr411 of HSA.23) The modification ratio was determined spectrophotometrically,23) indicating that 5.7% of the tyrosine residues were modified. These data suggest that about one out of 18 tyrosine residues in HSA had been modified. No structural changes in these chemically modified HSAs were detected, as evidenced by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and circular dichroism measurements.
Synthesis and Purification of Wild-Type HSA, W214A, R218A and Y411A MutantsThe recombinant DNA techniques used to produce recombinant wild-type HSA and single residue mutants, W214A, R218A and Y411A were essentially those described by Watanabe et al.24,25) SDS-PAGE and circular dichroism measurements indicated that there were no structural changes in the wild-type HSA and the mutant-HSAs. The wild-type HSA and the mutant-HSAs were stored at −20°C until used.
Statistical AnalysisStatistical analyses were performed using Dunnett’s test for multiple comparisons after one-way ANOVA. A probability value of p<0.05 was considered to be significant.
RESULTS
Identification of the Binding of Silibinin to Proteins in Human PlasmaTo identify the proteins in human plasma that bind silibinin, the binding experiments were conducted using isolated HSA, AAG and γ-globulins, at concentrations corresponding to those in human plasma (Fig. 2). Here, for the binding experiments, we choose a clinically relevant silibinin concentration at which therapeutic doses are administered.1,4,26–28) Ultrafiltration experiments revealed that the free fraction of silibinin in the HSA solution was comparable to that in human plasma. On the other hand, much higher free fractions were observed for AAG and γ-globulins solutions. These results indicate that silibinin mainly binds to HSA in human plasma.
Determination of the Binding Parameters of Silibinin for HSAThe binding parameters of silibinin for HSA were determined by means of ultrafiltration and equilibrium dialysis techniques. The binding parameters for a high-affinity binding site, n and K values, obtained by fitting to the binding equation, are summarized in Table 1. Data from both methods indicated that silibinin strongly binds to a single high affinity binding site on HSA. No differences were observed between the parameters obtained from ultrafiltration and equilibrium dialysis techniques.
Table 1. Binding Parameters Obtained by Ultrafiltration and Equilibrium Dialysis for Binding of Silibinin to HSA at pH 7.4 and 25°C
| Parameters | Ultrafiltration | Equilibrium dialysis |
|---|
| K(×105 M−1) | 4.81±0.22 | 4.45±0.17 |
| n | 1.02±0.05 | 0.98±0.07 |
The results are means±standard deviation (S.D.) (N=3).
Thermodynamic parameters for silibinin–HSA interactions were calculated by analyzing the temperature dependency of the association constant obtained by equilibrium dialysis. The van’t Hoff lines obtained by plotting ln K against 1/T were linear (data not shown), indicating that the ΔH values are constant over the experimental temperature range studied. As shown in Table 2, the formation of silibinin–HSA complexes is a spontaneous process, as evidenced by negative ΔG. Furthermore, the process is an exothermic reaction accompanied by a negative ΔH and ΔS. Consequently, binding processes is mainly driven by enthalpy.
Table 2. Thermodynamic Parameters for Silibinin–HSA Interaction at pH 7.4
| Temperature (K) | ΔG (kJ·mol−1) | ΔH (kJ·mol−1) | ΔS (J·K−1·mol−1) |
|---|
| 288 | −32.67±0.02 | −42.44±0.78 | −33.91±2.74 |
| 298 | −32.33±0.04 |
| 303 | −32.16±0.05 |
| 308 | −31.96±0.07 |
The results are means±S.D. (N=3).
To confirm the binding forces of silibinin to HSA, the effects of NaCl as an electrolyte and PLE as a nonionic detergent on the silibinin–HSA interaction were examined. PLE was added so that its concentration was below the critical micelle concentration (approx. 100 µM). The addition of NaCl and PLE to the medium increased the free fraction of silibinin in a concentration-dependent manner (Fig. 3).
Interaction between Silibinin and Sites I or II Marker Ligands on HSATo determine the site at which silibinin binds to HSA, mutual displacement between silibinin and typical site marker ligands (site I; warfarin, site II; ibuprofen) were analyzed using ultrafiltration techniques (Figs. 4A, B, a, b). The binding curve for silibinin in the presence of warfarin is close to the curve assuming competitive binding, and spontaneously, the binding curve of warfarin in the presence of silibinin is also close to the curve assuming competitive binding. In contrast, for silibinin–ibuprofen, almost no displacement was observed and the binding curve was close to the curve assuming independent binding. These data suggest that site I is the primary binding site of silibinin. Furthermore, mutual displacement between silibinin and other site I ligands, iodipamide and DNSA were also investigated (Figs. 4C, D, c, d). Silibinin was shown to interact with iodipamide in a competitive manner, but its binding to DNSA was in an independent manner.
Binding of Silibinin to HSA and Chemically Modified HSAsTo estimate the specific amino acid(s) that is involved in the binding of silibinin, the binding of free fractions of silibinin to chemically modified HSAs obtained from ultrafiltration data were compared to that for HSA (Fig. 5). The binding of warfarin and ibuprofen were decreased by the chemical modification of tryptophan and tyrosine residues on HSA, respectively. Similar to warfarin binding, the binding of silibinin to tryptophan-modified HSA was inhibited, whereas no effect was observed in the case where the tyrosine residue was modified. These data suggest that silibinin binds to the region close to the tryptophan residue in site I.
Binding of Silibinin to Wild-Type and Mutant HSAsSince Trp214 and Arg218 are known to be involved in the binding of site I ligands,29,30) silibinin binding was further characterized using mutant HSAs (W214A and R218A) (Fig. 6). The data obtained from ultrafiltration experiments showed that substituting Trp214 and Arg218 for a relatively small amino acid, alanine, caused a decrease in the binding of silibinin. In contrast, substituting Tyr411 which is located in the site II region24,25) for alanine (Y411A) had no effect on the binding of silibinin (Fig. 6). These data strongly suggest that silibinin binds to a region close to Trp214 and Arg218 in site I.
DISCUSSION
Recent evidence suggests that silymarin and its major constituent, silibinin, has therapeutic potential for the treatment of liver diseases such as hepatitis, hepatic cirrhosis, alcoholic liver disease and amatoxin mushroom poisoning,1,3,4,9) and cancers such as skin, prostate, breast, bladder and colorectal cancers.6–8,26) Furthermore, their preventive activities against gastrointestinal problems, nephropathy, cardio-plumonary problem have also been suggested.5) However, the fact that the protein binding of silibinin is not well understood poses limitations to its prospective clinical use. We carried out this study in order to address this issue, and have come to the following conclusions: 1) HSA is the main silibinin binding protein in human plasma; 2) The binding of silibinin to HSA appears to be assisted by multiple types of forces; 3) Silibinin and site I ligands, warfarin and iodipamide, displace each other; 4) Silibinin binds to the region close to Trp214 and Arg218 in site I of HSA. These findings led us to develop a detailed understanding of the protein binding of silibinin and a precise prediction of the pharmacokinetics and therapeutic effects of silibinin and co-administered drugs in patients.
Our results from ultrafiltration techniques using human plasma and its component proteins indicated, for the first time, that silibinin preferentially associates with HSA but not AAG or γ-globulins (Fig. 2). The binding parameters for silibinin to HSA showed that silibinin forms a strong 1 : 1 complex with HSA (Table 1). In order to characterize the binding mode of silibinin to HSA, a thermodynamic analysis was performed. As shown in Table 2, all of the measured thermodynamic parameters (ΔG, ΔH, ΔS) were negative. These results indicate that the formation of a silibinin–HSA complex is a spontaneous and exothermic process and that it is driven by enthalpy. Ross and Subramanian31) characterized the sign and magnitude of the thermodynamic parameters for various types of ligand–protein interactions. According to their characterization, the negative ΔH and ΔS values observed in silibinin–HSA interactions can largely be atributed to van der Waals forces and/or hydrogen bond formation. These are similar to the interactions of other polyphenols with albumins.32–35) Maiti et al.17) indicated an initial hydrophobic association followed by electrostatic interactions as well as van der Waals interactions and hydrogen bonding in the subsequent interacting complex based on thermodynamic parameters using association constants obtained from fluorescence quenching method (ΔG; −29.83~−28.35 kJ/mol, ΔH; −6.76 kJ/mol, ΔS; 73.69 J/mol/K). Our conclusion is somewhat different from theirs, possibly due to the differences in the methodology used. However, hydrophobic and electrostatic interactions cannot be excluded, even from our data, because such interactions may be involved within the dynamic process until complex formation between silibinin and HSA occurs.31,35) Indeed, the free fraction of silibinin was dependent on the ionic strength of the medium and the concentration of non-ionic detergent (Fig. 3), suggesting that electrostatic and hydrophobic interactions are also involved as a driving force for binding. Thus, not a single force but multiple forces, including hydrogen bonds, van der Waals forces, hydrophobic and electrostatic interactions appear to play important roles in the binding of silibinin to HSA.
The binding site of silibinin on HSA was determined by analyzing the interaction mode between silibinin and site-marker ligands (Fig. 4). Competitive interactions were observed between silibinin and warfarin on HSA (Fig. 4A, a), while the binding of silibinin to HSA is likely to be independent of ibuprofen binding (Fig. 4B, b). These findings strongly suggest that, at therapeutic concentrations, silibinin preferentially binds to site I where warfarin also binds, but not to site II, where ibuprofen binds. Fehske et al. previously identified tryptophan and tyrosine residues as part of sites I and II, respectively, based on binding studies using chemically modified HSAs.28,36) In the present study, we examined the binding of silibinin to chemically modified HSAs to collect further evidence to show that silibinin binds to site I and to confirm whether Trp214, one of the residues in the site I region, is involved in silibinin binding. As described in Materials and Methods, Trp214 and Tyr411 can be selectively modified by HNB and p-nitrophenyl anthranilate, respectively. Therefore, the results for the binding of silibinin to these modified HSAs (Fig. 5) suggest that silibinin binding occurs in a region close to Trp214 in site I, but not Tyr411 in site II. Warfarin and silibinin binding were similarly decreased by chemical modification (Figs. 5A, B), suggesting the presence of overlapping of warfarin- and silibinin-binding regions in site I. These data were supported by the fact that the binding of silibinin to HSA was significantly decreased when Trp214 in site I was mutated to alanine (W214A, Fig. 6). In addition, similar to the binding of typical site I drugs, including warfarin,24) the binding of silibinin to HSA was also decreased by the mutation of Arg218 to alanine (R218A, Fig. 6). It therefore appears that Arg218 as well as Trp214 are located in close proximity to binding region of silibinin in site I.
Kragh-Hansen reported that site I is “a large and flexible region” based on the diversity of ligands that interact and the apparent ability to accommodate more than one of them at a time.19) Crystallographic studies also directed at HSA-site I drug complexes demonstrated that site I is larger than site II and that site I drugs occupy different regions of the binding pocket of subdomain IIA.29,36) The interior of site I is predominantly apolar, but contains two clusters of polar residues (Tyr150, His242, Arg257 as an inner cluster and Lys195, Lys199, Arg218, Arg222 as an outer cluster).29) Warfarin participates in hydrogen bonding with Tyr150 His242 and Arg222 in the center of the site I pocket, where iodipamide, a larger molecule (MW: 1140 Da), occupies the wider region from the back-end to front in site I with interacting with Arg257, Trp214, Lys198.29) We previously reported that site I is not a simple binding region but is rather complex and is comprised of three subsites, namely, Ia, Ib and Ic.37) Warfarin binds to subsite Ia, and iodipamide binds to a wider region, subsites Ia and Ic.37) Competitive interactions of silibinin–warfarin and silibinin–iodipamide and independent binding observed in the interaction between silibinin and DNSA, subsite Ib ligands,37) indicate that silibinin binds to subsite Ia and/or Ic in site I. Similar to iodipamide, silibinin which has relatively large molecular size (Fig. 1, MW: 482 Da) may occupy both Ia and Ic. Maiti et al., through molecular docking studies, suggested that silibinin lies within hydrogen bonding distance of Trp214 and Asp451 of subdomains IIA and IIIA, respectively.17) Although our results did not show an interaction of silibinin with subdomain IIIA, the present data may also indicate that silibinin occupies the region at the entrance of the site I pocket, which faces subdomain IIIA as iodipamide.29) Flavonoids including its glycoside substituents have been shown to bind site I.32,38–40) In flavonoid-site I interactions, hydrogen bonding between the 7-OH of the benzopyrone moiety and Arg222 or 5-OH and Arg257 have been proposed as warfarin–site I and iodipamide–site I interactions.29,40) Like the relatively large flavonoid, quercetin,40,41) the benzopyrone moiety of silibinin may be located within the pocket in site I, while the other parts may protrude toward the interface between subdomains IIA and IIIA. Further studies directed at identifying the exact silibinin binding site and the amino acids other than Trp214 and Arg218 that contribute to silibinin binding should be conducted by binding studies with the other mutants or a crystallographic study.
Silymarin and its main constituent, silibinin are generally considered to be safe because their adverse effects are rare and are limited to nausea, headache, joint pain, itching and mild laxative symptoms.1,4,9) Therefore, relatively high doses of silibinin (up to 480 mg/d) have been orally administered in clinical trials.4) Furthermore, dietary supplements that show a high bioavailability (e.g., Siliphos®, complex of silibinin with phosphatidylcholine) are currently being marketed world-wide.27,42) As a dietary supplement, approximately 50–60 mg twice to three times daily is suggested. However, overdoses by patients who abuse such supplements can cause unexpected accumulations, thereby eliciting interactions with co-administered drugs. Furthermore, considering the selective binding of silibinin to subsites Ia and/or Ic of site I, the pharmacokinetics and pharmacodynamics of drugs that bind to these regions (e.g., warfarin or iodipamide) may be modified by silibinin. Thus, changes in drug efficacy and the appearance of adverse effects should be monitored carefully when silibinin is administered in conjunction with site I drugs. Modification of the silibinin binding by co-administered drugs or endogenous compounds that accumulate at abnormally high levels in diseased states also should be considered in predicitng its pharmacological action. Warfarin, iodipamide, furosemide or bucolome which all strongly bind to site I43) may displace silibinin. 3-Carboxy-4-methyl-5-propyl-2-furanpropionic acid (CMPF) which accumulates in patients with renal disease also exerts inhibitory effects on site I ligand bindings.44) The unbound fraction of the site I drug, phenytoin, in patients with hepatitis or cirrhosis is known to correlate with the plasma concentration of bilirubin that also binds to site I.45) Changes in the concentrations of plasma proteins can also affect silibinin binding, although these effects do not appear to be site-specific. In the diseased state, the plasma concentration of HSA is generally decreased, resulting in decreased silibinin binding. While AAG and γ-globulins are minor contributors to the binding of silibinin to proteins in plasma (Fig. 2), binding to these proteins might need to be considered in cases of inflammation or infection, since the plasma concentrations of AAG and γ-globulins typically increase significantly under these conditions, compared to normal.46–50) Thus, the findings regarding the binding of silibinin to plasma protein reported in this study will permit a more relevant assessment of the pharmacokinetics and pharmacodynamics of silibinin and co-administered drugs in various clinical situations.
CONCLUSION
In this study, we attempted to clarify the mechanism and the site at which silibinin binds to HSA. Silibinin interacts with subsites Ia and/or Ic of site I on HSA with assistance by multiple forces, including hydrogen bonds, van der Waals forces, hydrophobic and electrostatic interactions. Trp214 and Arg218 were identified as amino acid residues that are located close to the binding site for silibinin. Such a detailed analysis of silibinin–HSA interactions provides valuable information in terms of our understanding of the pharmacokinetics and the pharmacological effects of silibinin and related co-administered drugs. Furthermore, the findings presented herein will also be useful when the multiple pharmacological actions of silibinin are clinically applied for the treatment of a variety of diseases.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Wellington K, Jarvis B. Silymarin: a review of its clinical properties in the management of hepatic disorders. BioDrugs, 15, 465–489 (2001).
- 2) Kroll DJ, Shaw HS, Oberlies NH. Milk thistle nomenclature: why it matters in cancer research and pharmacokinetic studies. Integr. Cancer Ther., 6, 110–119 (2007).
- 3) Abenavoli L, Capasso R, Milic N, Capasso F. Milk thistle in liver diseases: past, present, future. Phytother. Res., 24, 1423–1432 (2010).
- 4) Loguercio C, Festi D. Silybin and the liver: from basic research to clinical practice. World J. Gastroenterol., 17, 2288–2301 (2011).
- 5) Kren V, Walterova D. Silybin and silymarin—new effects and applications. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub., 149, 29–41 (2005).
- 6) Kaur M, Agarwal R. Silymarin and epithelial cancer chemoprevention: how close we are to bedside? Toxicol. Appl. Pharmacol., 224, 350–359 (2007).
- 7) Deep G, Agarwal R. Antimetastatic efficacy of silibinin: molecular mechanisms and therapeutic potential against cancer. Cancer Metastasis Rev., 29, 447–463 (2010).
- 8) Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer potential of silymarin: from bench to bed side. Anticancer Res., 26 (6B), 4457–4498 (2006).
- 9) Singh RP, Agarwal R. Prostate cancer chemoprevention by silibinin: bench to bedside. Mol. Carcinog., 45, 436–442 (2006).
- 10) Peters T. All About Albumin: Biochemistry, Genetics, and Medical Applications. Academic Press, San Diego, California (1995).
- 11) Kragh-Hansen U, Chuang VT, Otagiri M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull., 25, 695–704 (2002).
- 12) Sudlow G, Birkett DJ, Wade DN. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol., 11, 824–832 (1975).
- 13) Sudlow G, Birkett DJ, Wade DN. Further characterization of specific drug binding sites on human serum albumin. Mol. Pharmacol., 12, 1052–1061 (1976).
- 14) He XM, Carter DC. Atomic structure and chemistry of human serum albumin. Nature, 358, 209–215 (1992).
- 15) Carter DC, Ho JX. Structure of serum albumin. Adv. Protein Chem., 45, 153–203 (1994).
- 16) Petitpas I, Bhattacharya AA, Twine S, East M, Curry S. Crystal structure analysis of warfarin binding to human serum albumin: anatomy of drug site I. J. Biol. Chem., 276, 22804–22809 (2001).
- 17) Maiti TK, Ghosh KS, Samanta A, Dasgupta S. The interaction of silibinin with human serum albumin: A spectroscopic investigation. J. Photochem. Photobiol. Chem., 194, 297–307 (2008).
- 18) Giacomini KM, Wong FM, Tozer TN. Correction for Volume Shift during Equilibrium Dialysis by Measurement of Protein Concentration. Pharm. Res., 1, 179–181 (1984).
- 19) Kragh-Hansen U. Evidence for a large and flexible region of human serum albumin possessing high affinity binding sites for salicylate, warfarin, and other ligands. Mol. Pharmacol., 34, 160–171 (1988).
- 20) Kragh-Hansen U. Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev., 33, 17–53 (1981).
- 21) Pedersen AO, Honore B, Brodersen R. Thermodynamic parameters for binding of fatty acids to human serum albumin. Eur. J. Biochem., 190, 497–502 (1990).
- 22) Fehske KJ, Muller WE, Wollert U. The modification of the lone tryptophan residue in human serum albumin by 2-hydroxy-5-nitrobenzyl bromide. Characterization of the modified protein and the binding of L-tryptophan and benzodiazepines to the tryptophan-modified albumin. Hoppe Seylers Z. Physiol. Chem., 359, 709–717 (1978).
- 23) Hagag N, Birnbaum ER, Darnall DW. Resonance energy transfer between cysteine-34, tryptophan-214, and tyrosine-411 of human serum albumin. Biochemistry, 22, 2420–2427 (1983).
- 24) Watanabe H, Kragh-Hansen U, Tanase S, Nakajou K, Mitarai M, Iwao Y, Maruyama T, Otagiri M. Conformational stability and warfarin-binding properties of human serum albumin studied by recombinant mutants. Biochem. J., 357, 269–274 (2001).
- 25) Watanabe H, Tanase S, Nakajou K, Maruyama T, Kragh-Hansen U, Otagiri M. Role of arg-410 and tyr-411 in human serum albumin for ligand binding and esterase-like activity. Biochem. J., 349, 813–819 (2000).
- 26) Hoh C, Boocock D, Marczylo T, Singh R, Berry DP, Dennison AR, Hemingway D, Miller A, West K, Euden S, Garcea G, Farmer PB, Steward WP, Gescher AJ. Pilot study of oral silibinin, a putative chemopreventive agent, in colorectal cancer patients: silibinin levels in plasma, colorectum, and liver and their pharmacodynamic consequences. Clin. Cancer Res., 12, 2944–2950 (2006).
- 27) Kidd P, Head K. A review of the bioavailability and clinical efficacy of milk thistle phytosome: a silybin–phosphatidylcholine complex (Siliphos). Altern. Med. Rev., 10, 193–203 (2005).
- 28) Zhu HJ, Brinda BJ, Chavin KD, Bernstein HJ, Patrick KS, Markowitz JS. An assessment of pharmacokinetics and antioxidant activity of free silymarin flavonolignans in healthy volunteers: a dose escalation study. Drug Metab. Dispos., 41, 1679–1685 (2013).
- 29) Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol., 353, 38–52 (2005).
- 30) Sato H, Chuang VT, Yamasaki K, Yamaotsu N, Watanabe H, Nagumo K, Anraku M, Kadowaki D, Ishima Y, Hirono S, Otagiri M, Maruyama T. Differential effects of methoxy group on the interaction of curcuminoids with two major ligand binding sites of human serum albumin. PLoS ONE, 9, e87919 (2014).
- 31) Ross PD, Subramanian S. Thermodynamics of protein association reactions: forces contributing to stability. Biochemistry, 20, 3096–3102 (1981).
- 32) Dai J, Zou T, Wang L, Zhang Y, Liu Y. Investigation of the interaction between quercetin and human serum albumin by multiple spectra, electrochemical impedance spectra and molecular modeling. Luminescence, 29, 1154–1161 (2014).
- 33) Xiao JB, Chen XQ, Jiang XY, Hilczer M, Tachiya M. Probing the interaction of trans-resveratrol with bovine serum albumin: a fluorescence quenching study with Tachiya model. J. Fluoresc., 18, 671–678 (2008).
- 34) Frazier RA, Papadopoulou A, Green RJ. Isothermal titration calorimetry study of epicatechin binding to serum albumin. J. Pharm. Biomed. Anal., 41, 1602–1605 (2006).
- 35) Zhang X, Li L, Xu Z, Liang Z, Su J, Huang J, Li B. Investigation of the interaction of naringin palmitate with bovine serum albumin: spectroscopic analysis and molecular docking. PLoS ONE, 8, e59106 (2013).
- 36) Zhu L, Yang F, Chen L, Meehan EJ, Huang M. A new drug binding subsite on human serum albumin and drug–drug interaction studied by X-ray crystallography. J. Struct. Biol., 162, 40–49 (2008).
- 37) Yamasaki K, Maruyama T, Kragh-Hansen U, Otagiri M. Characterization of site I on human serum albumin: concept about the structure of a drug binding site. Biochim. Biophys. Acta, 1295, 147–157 (1996).
- 38) Bolli A, Marino M, Rimbach G, Fanali G, Fasano M, Ascenzi P. Flavonoid binding to human serum albumin. Biochem. Biophys. Res. Commun., 398, 444–449 (2010).
- 39) Boulton DW, Walle UK, Walle T. Extensive binding of the bioflavonoid quercetin to human plasma proteins. J. Pharm. Pharmacol., 50, 243–249 (1998).
- 40) Ionescu S, Matei I, Tablet C, Hillebrand M. New insights on flavonoid-serum albumin interactions from concerted spectroscopic methods and molecular modeling. Curr. Drug Metab., 14, 474–490 (2013).
- 41) Zsila F, Bikadi Z, Simonyi M. Probing the binding of the flavonoid, quercetin to human serum albumin by circular dichroism, electronic absorption spectroscopy and molecular modelling methods. Biochem. Pharmacol., 65, 447–456 (2003).
- 42) Kidd PM. Bioavailability and activity of phytosome complexes from botanical polyphenols: the silymarin, curcumin, green tea, and grape seed extracts. Altern. Med. Rev., 14, 226–246 (2009).
- 43) Yamasaki K, Chuang VT, Maruyama T, Otagiri M. Albumin–drug interaction and its clinical implication. Biochim. Biophys. Acta, 1830, 5435–5443 (2013).
- 44) Sakai T, Takadate A, Otagiri M. Characterization of binding site of uremic toxins on human serum albumin. Biol. Pharm. Bull., 18, 1755–1761 (1995).
- 45) Hooper WD, Bochner F, Eadie MJ, Tyrer JH. Plasma protein binding of diphenylhydantoin. Effects of sex hormones, renal and hepatic disease. Clin. Pharmacol. Ther., 15, 276–282 (1974).
- 46) Kremer JM, Wilting J, Janssen LH. Drug binding to human alpha-1-acid glycoprotein in health and disease. Pharmacol. Rev., 40, 1–47 (1988).
- 47) Chuang VT, Otagiri M. Photoaffinity labeling of plasma proteins. Molecules, 18, 13831–13859 (2013).
- 48) Hunziker L, Recher M, Macpherson AJ, Ciurea A, Freigang S, Hengartner H, Zinkernagel RM. Hypergammaglobulinemia and autoantibody induction mechanisms in viral infections. Nat. Immunol., 4, 343–349 (2003).
- 49) Maher FT, Snell AM, Mann FD. Turbidimetric estimation of serum colloids in the differential diagnosis of hepatobiliary disease. Gastroenterology, 12, 394–408 (1949).
- 50) Zitrin CM, Lincoln EM, Saifer A, Lewkowicz S. Serum gamma globulin in childhood tuberculosis. Am. Rev. Tuberc., 74, 15–28 (1956).





