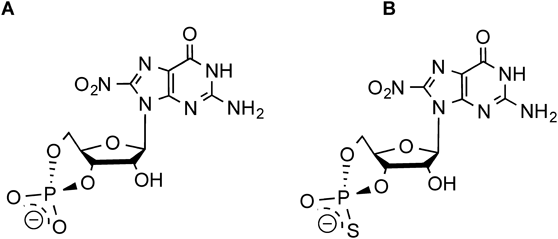
Abstract
Guanosine 3′,5′-cyclic monophosphate (cGMP)-dependent protein kinases (PKG) are kinases regulating diverse physiological functions including vascular smooth muscle relaxation, neuronal synaptic plasticity, and platelet activities. Certain PKG inhibitors, such as Rp-diastereomers of derivatives of guanosine 3′,5′-cyclic monophosphorothioate (Rp-cGMPS), have been designed and used to study PKG-regulated cell signaling. 8-Nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP) is an endogenous cGMP derivative formed as a result of excess production of reactive oxygen species and nitric oxide. 8-Nitro-cGMP causes persistent activation of PKG1α through covalent attachment of cGMP moieties to cysteine residues of the enzyme (i.e., the process called protein S-guanylation). In this study, we synthesized a nitrated analogue of Rp-cGMPS, 8-nitroguanosine 3′,5′-cyclic monophosphorothioate Rp-isomer (Rp-8-nitro-cGMPS), and investigated its effects on PKG1α activity. We synthesized Rp-8-nitro-cGMPS by reacting Rp-8-bromoguanosine 3′,5′-cyclic monophosphorothioate (Rp-8-bromo-cGMPS) with sodium nitrite. Rp-8-Nitro-cGMPS reacted with the thiol compounds cysteine and glutathione to form Rp-8-thioalkoxy-cGMPS adducts to a similar extent as did 8-nitro-cGMP. As an important finding, a protein S-guanylation-like modification was clearly observed, by using Western blotting, in the reaction between recombinant PKG1α and Rp-8-nitro-cGMPS. Rp-8-Nitro-cGMPS inhibited PKG1α activity with an inhibitory constant of 22 µM in a competitive manner. An organ bath assay with mouse aorta demonstrated that Rp-8-nitro-cGMPS inhibited vascular relaxation induced by acetylcholine or 8-bromo-cGMP more than Rp-8-bromo-cGMPS did. These findings suggest that Rp-8-nitro-cGMPS inhibits PKG through induction of an S-guanylation-like modification by attaching the Rp-cGMPS moiety to the enzyme. Additional study is warranted to explore the potential application of Rp-8-nitro-cGMPS to biochemical and therapeutic research involving PKG1α activation.
Guanosine 3′,5′-cyclic monophosphate (cGMP)-dependent protein kinase, or protein kinase G (PKG), is a serine/threonine kinase that regulates diverse physiological functions such as vascular smooth muscle relaxation, neuronal synaptic plasticity, and platelet activities.1) Two different PKG genes were identified in eukaryotes: prkg1, coding for PKG1, and prkg2, coding for PKG2.2) Alternative splicing leads to expression of two PKG1 isoforms, PKG1α and PKG1β. PKG1α is expressed in several tissues, mostly lung, cerebellum, and heart, whereas PKG1β is expressed predominantly in platelets and hippocampus.3) Both PKG1α and PKG1β occur in vascular smooth muscle cells, uterus, gastrointestinal tract, kidney, and trachea.3) Tissue distribution of PKG2 was reported in the kidney, intestine, lung, brain, and chondrocytes.3) PKG is present in cells in a resting (inactive) state in the absence of cGMP.1) Binding of cGMP to the regulatory domain of the enzyme causes the dissociation of the N-terminal autoinhibitory domain from the catalytic center, initiating the phosphorylation of serine/threonine residues in target proteins.
A number of PKG inhibitors have been designed and used to study the fundamental roles of PKG in cell signaling.4,5) The Rp-diastereomers of analogues of guanosine 3′,5′-cyclic monophosphorothioate (Rp-cGMPS) have long been studied as competitive inhibitors of PKG1α.4,5) They bind to PKG but apparently do not evoke conformational changes of the enzyme required for its activation.6) Rp-8-Bromoguanosine 3′,5′-cyclic monophosphorothioate (Rp-8-bromo-cGMPS) is a widely used PKG inhibitor in signal transduction research. Accumulating evidence suggested that PKG inhibition may be a potential target in the treatment of certain diseases such as bacterial toxin-induced diarrhea and cancers of the lung, colon, breast, and ovary.4,5)
We discovered endogenous formation of the nitrated cGMP derivative 8-nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP) under conditions associated with excess production of both nitric oxide (NO) and reactive oxygen species in mammalian cells7–17) as well as in plants.18,19) Biochemical analyses revealed that 8-nitro-cGMP can activate PKG1α to a similar extent as native cGMP can.9,12) Although native cGMP is degraded by phosphodiesterase to form guanosine 5′-monophosphate, 8-nitro-cGMP was resistant to phosphodiesterase-mediated degradation.9) Furthermore, 8-nitro-cGMP covalently bound to cysteine residues in PKG1α through the formation of cysteinyl-cGMP adducts in the enzyme (i.e., the process called protein S-guanylation).12) Two S-guanylation sites were identified in PKG1α: Cys42, near the autoinhibitory domain, and Cys195, located at the high-affinity cGMP-binding site.12) That S-guanylation at Cys195 leads to persistent activation of the enzyme is interesting to note.12)
As mentioned above, nitration of the C8 carbon in cGMP makes the carbon electrophilic in a reaction with nucleophilic cysteine residues of PKG.12) Like 8-nitro-cGMP, a nitrated analogue of Rp-cGMPS may covalently bind to PKG through an S-guanylation-like modification, which would result in fixation of the Rp-cGMPS moiety to the regulatory domain in the enzyme. In this study, we synthesized Rp-8-nitroguanosine 3′,5′-cyclic monophosphorothioate (Rp-8-nitro-cGMPS) (Fig. 1). We determined the electrophilic property of the compound in terms of the ability of the compound to induce an S-guanylation-like reaction. We studied the effects of Rp-8-nitro-cGMPS on PKG1α activity by using the recombinant enzyme in vitro as well as in an organ bath system. Our data suggested that Rp-8-nitro-cGMPS demonstrated a more potent inhibition of vascular relaxation induced by acetylcholine and by 8-bromoguanosine 3′,5′-cyclic monophosphate (8-bromo-cGMP) in the organ bath system than did the widely used PKG inhibitor Rp-8-bromo-cGMPS. These findings warrant continued investigation of the use of Rp-8-nitro-cGMPS as a biochemical tool in signal transduction and as a potent inhibitor for therapeutic purposes.
MATERIALS AND METHODS
MaterialsRp-8-Bromo-cGMPS was purchased from Biolog Life Science Institute (Bremen, Germany). 8-Bromo-cGMP was obtained from Sigma-Aldrich (Saint Louis, MO, U.S.A.). 8-Nitro-cGMP was synthesized as reported previously.7,9) Recombinant human PKG1α (N-terminal 6His-tagged, expressed by baculovirus in the Sf21 insect cell line) was from Merck Millipore (Billerica, MA, U.S.A.). A NO donor, 1-hydroxy-2-oxo-3-(N-ethyl-2-aminoethyl)-3-ethyl-1-triazene (NOC 12) was from Dojindo (Kumamoto, Japan). Other reagents were of the highest grade commercially available and were used without additional purification.
Synthesis of Rp-8-Nitro-cGMPSRp-8-Nitro-cGMPS was synthesized from Rp-8-bromo-cGMPS by modifying our nitration reaction method for 8-bromo-cGMP.7,9) In brief, 80 mM Rp-8-bromo-cGMPS was reacted with 300 mM sodium nitrite in dimethyl sulfoxide (DMSO) containing 23 mM HCl at 70°C for 3 d. To remove sodium nitrite and DMSO, the reaction mixture was diluted with 0.1% formic acid and then purified by using an octadecylsilane (ODS) solid phase extraction column (Sep-Pak C18 12 cc Vac Cartridge; Waters, Milford, MA, U.S.A.) according to the manufacturer’s instructions. Rp-8-Nitro-cGMPS was purified by means of HPLC with a semi-preparative reverse-phase (RP) column (Capcell Pak C18 column, 20.0 mm i.d.×150 mm; Shiseido, Tokyo, Japan) at 40°C. Semi-preparative RP-HPLC was performed by using the Waters e2695 series HPLC system with the UV detector set at 400 nm. Mobile phases A (10 mM sodium borate pH 9.0 containing 50 mM NaCl) and B (100% methanol) were used with a linear gradient of B of 0 to 20% for 100 min and a flow rate of 3 mL/min. Purified Rp-8-nitro-cGMPS was loaded onto the ODS solid phase extraction column (Sep-Pak C18 6 cc Vac Cartridge; Waters) to change the solvent and was then eluted with 60% aqueous methanol. Eluted samples were dried in vacuo and then dissolved in ultrapure water. Concentrations of Rp-8-nitro-cGMPS solutions were determined by measuring the absorption at 390 nm in neutral buffer, for which we assumed that Rp-8-nitro-cGMPS has a molecular coefficient equivalent to that of 8-nitro-cGMP.7)
HPLC Analysis of Cyclic Nucleotide InhibitorsRp-8-Bromo-cGMPS and Rp-8-nitro-cGMPS were injected onto an analytical RP column (YMC-Triart Plus C18, 4.6 mm i.d.×150 mm; YMC Co., Ltd., Kyoto, Japan). Mobile phases A (H2O+0.1% formic acid) and B (acetonitrile) were used with a linear gradient of B of 1 to 15% for 20 min and a flow rate of 0.8 mL/min, with the UV detector set at 210, 254, and 400 nm. For analysis of the reaction between Rp-8-nitro-cGMPS and cysteine, the linear gradient of B was changed to 5 to 20% for 20 min.
LC-MSMass spectrometric identification of cyclic nucleotide inhibitors including Rp-8-nitro-cGMPS and Rp-8-cystenyl-cGMPS was performed by using Agilent 6460 Triple Quadrupole LC/MS (Agilent Technologies, Santa Clara, CA, U.S.A.). LC conditions used were as follows: column, YMC-Triart Plus C18 (2.1 mm i.d.×50 mm) (YMC Co., Ltd.); column temperature, 45°C; injection volume, 10 µL; mobile phases, A: H2O+0.1% formic acid and B: acetonitrile (concentration), 0 min: 1%, 12 min: 15%, 12.5 min: 1%, 15 min: 1%; and flow rate, 0.2 mL/min. MS scan parameters were as follows: scan range (m/z), 100–500; fragmentor voltage, 130 V; polarity, negative. Multiple reaction monitoring (MRM) analysis was performed for quantification of Rp-8-bromo-cGMPS and Rp-8-nitro-cGMPS in mouse aortic rings with the MRM parameters as follows: for Rp-8-bromo-cGMPS: precursor ion (m/z), 437.9; product ion (m/z), 227.8; fragmentor voltage, 130 V; collision energy, 25 V; polarity, negative; and for Rp-8-nitro-cGMPS: precursor ion (m/z), 405; product ion (m/z), 194.9; fragmentor voltage, 130 V; collision energy, 21 V; polarity, negative. The inhibitors were extracted from mouse aortic rings by using the protocol reported previously.12)
Kinetic Analysis of the Reaction between Rp-8-Nitro-cGMPS and ThiolsRp-8-Nitro-cGMPS (20 µM) was reacted with 10 mM thiols (cysteine or glutathione) in 200 mM sodium phosphate buffer (pH 7.4) at 37°C. Changes in absorbance of the resultant solutions were determined by using the BioSpectrometer Kinetic spectrophotometer (Eppendorf, Hamburg, Germany). Absorbance at 390 nm was plotted against incubation time. Apparent second-order rate constants were determined from the slope of the plot of incubation time versus log(A−A0), where A0 is the initial absorbance.7)
Protein S-Guanylation-Like Modification of PKG1α by Rp-8-Nitro-cGMPSRecombinant PKG1α (100 ng) was reacted with 8-nitro-cGMP or Rp-8-nitro-cGMPS at concentrations of 1, 10, or 30 µM in 100 mM Tris–HCl (pH 7.4) at 37°C for 120 min. The resultant protein preparations were subjected to Western blotting as described below.
Western BlottingProteins were separated by using 10 or 12% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and were then transferred to a polyvinylidene fluoride membrane. Blots were blocked with 5% nonfat skim milk followed by incubation with one of the following antibodies: anti-S-guanylation,7,8,16) anti-PKG (Santa Cruz Biotechnology, Dallas, TX, U.S.A.), anti-phospho-VASP [at serine 239; Ser239] (Cell Signaling Technology, Danvers, MA, U.S.A.) or anti-vasodilator-associated stimulated protein (VASP) (Enzo Life Sciences, Farmingdale, NY, U.S.A.). After incubation of the samples with adequate secondary antibodies conjugated with horseradish peroxidase, immunoreactive bands were detected by using a chemiluminescence reagent (ECL Plus; GE Healthcare Bio-Sciences Corp., Piscataway, NJ, U.S.A.) and a luminescent image analyzer (LAS1000UV; FUJIFILM, Tokyo, Japan).
In Vitro Kinase AssayPKG activity was measured as reported previously.12) In brief, 1 µg/mL PKG1α (total 10 ng) was incubated with Rp-8-bromo-cGMP or Rp-8-nitro-cGMP at concentrations of 1, 10, or 100 µM for 90 min before the kinase experiments. PKG1α reaction mixtures with inhibitors were then used as kinase mixes during incubation with cGMP at different concentrations (0, 1, 10, or 100 µM) in 20 mM Tris–HCl (pH 7.4) containing 16 mM MgCl2, 10 mM dithiothreitol, 0.1 mM ATP, and 60 µM Glasstide (Merck Millipore) at 37°C for 30 min. After the incubation, kinase activity was determined with the ADP-Glo Kinase assay kit (Promega, Madison, WI, U.S.A.) according to the manufacturer’s protocol. Dixon plot analyses were performed by using the Graphpad Prism Program to determine the inhibitory constants (Ki) for both Rp-8-bromo-cGMPS and Rp-8-nitro-cGMPS.
Organ Bath AssayThe animals were maintained and the experiments were conducted according to the guidelines in the Laboratory Protocol of Animal Handling, Kumamoto University Graduate School of Medical Sciences. Eight-week-old male C57BL/6J mice (Kyudo Co., Ltd., Saga, Japan) were euthanized by inhalation of diethylether and subjected to cervical dislocation, and the thoracic aortas were immediately excised. Vascular rings (5 mm long) thus obtained were mounted in a tension myograph (model MTOB-2Z; Lab Support, Suita, Japan) stretched to the optimal pretension conditions of 1.0 g. The aortic rings were bathed in Krebs–Henseleit buffer (118.4 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3, and 10 mM glucose, pH 7.6), maintained at 37°C, and gassed with 95% O2 and 5% CO2. The tone of the aortic vessel specimens was measured as described earlier.12) The aortic rings were treated first with PKG1α inhibitors (100 µM Rp-8-bromo-cGMPS or Rp-8-nitro-cGMPS) or they received no treatment. Then 100% contraction of vessels was achieved by treating the vessels with phenylephrine (1 µM) in Hanks’ balanced solution for 90 min. The vascular relaxation responses induced by acetylcholine and 8-bromo-cGMP were then recorded with a force transducer data acquisition system (PowerLab, Chart v5; ADInstruments, Colorado Springs, CO, U.S.A.).
Tissue Distribution of Rp-8-Nitro-cGMPSAortic rings from mice were treated with 100 µM Rp-8-nitro-cGMPS or Rp-8-bromo-cGMPS in Krebs–Henseleit buffer for 90 min at 37°C. The rings were then washed twice with phosphate-buffered saline (PBS) and were suspended in ice-cold methanol containing 2% acetic acid. Tissue suspensions thus obtained were homogenized by means of the TissueRuptor (Qiagen, Hilden, Germany) until the aortic vessels were completely homogenized. After centrifugation, supernatants were subjected to solid phase purification of cyclic nucleotide derivatives with use of the Oasis WAX cartridge (Waters) as previously described.12) Rp-8-Nitro-cGMPS and Rp-8-bromo-cGMPS extracted from aortic vessels were quantified by means of LC-electrospray ionization-MS/MS as described above.
Phosphorylation of VASP in Rat C6 Glioma CellsPhosphorylation of VASP at Ser239 was analyzed as an index of PKG activation.20,21) Rat C6 glima cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) that was supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. C6 cells (5×104 cells) were treated with NOC 12 to activate NO/cGMP/PKG signaling pathway that leads to enhanced VASP phosphorylation. To test the effects of PKG inhibitors on VASP phosphorylation, cells were incubated in the presence or absence of 100 µM of Rp-8-nitro-cGMPS or Rp-8-bromo-cGMPS for 4 h, followed by treating with 10 µM NOC 12 for 30 min. Cells were then washed with PBS and lysed with SDS sample buffer. Lysates were boiled at 95°C for 3 min and then subjected to Western blotting for phospho- and total-VASP.
Statistical AnalysisAll data are expressed as the mean±standard deviation (S.D.). Data for each experiment were acquired from at least three independent experiments. Statistical analyses were performed by using Student’s t-test, with the level of significance set at p<0.05.
RESULTS
Synthesis of Rp-8-Nitro-cGMPSAfter incubation of Rp-8-bromo-cGMPS with sodium nitrite in DMSO, the reaction solution became yellowish. RP-HPLC showed a new peak that eluted slower than did Rp-8-bromo-cGMPS. We collected the fraction containing the new peak by using a semi-preparative RP-HPLC column. After purification, the collected sample had a single RP-HPLC peak, without detectable contamination with Rp-8-bromo-cGMPS (Fig. 2A, Supplementary Fig. S1). UV spectrometry revealed that the compound had a maximum absorption at 395 nm (Fig. 2B). This UV spectrum was quite similar to that of 8-nitro-cGMP.7) MS strongly suggested that the compound was identical to Rp-8-nitro-cGMPS (Fig. 2C).
Reactions of Rp-8-Nitro-cGMPS with Low-Molecular-Weight ThiolsWe investigated the electrophilic property of Rp-8-nitro-cGMPS by means of its reactivity with thiols as biological nucleophiles. In the reaction between Rp-8-nitro-cGMPS and cysteine in neutral aqueous buffer, the absorption at 395 nm, which is characteristic of the 8-nitroguanine moiety, decreased in a time-dependent manner (Fig. 3A). After incubation of Rp-8-nitro-cGMPS with cysteine for 120 min, a peak corresponding to Rp-8-nitro-cGMPS almost completely disappeared and a new peak appeared in the RP-HPLC separation (Fig. 3B). Negative mode MS showed that the molecular mass of the new peak was 478.8 ([M−H+]–), which suggested the formation of Rp-8-cysteinyl-cGMPS (Fig. 3C). As Table 1 shows, the rate constants for the reaction between Rp-8-nitro-cGMPS and thiols (cysteine, glutathione) were almost identical to those for 8-nitro-cGMP and thiols, which suggested that Rp-8-nitro-cGMPS possesses electrophilicity comparable to that of 8-nitro-cGMP.
Table 1. Rate Constants for the Reaction of 8-Nitro-cGMP and Rp-8-Nitro-cGMPS with Low-Molecular-Weight Thiols
| Thiol | Reaction rate constant (M−1·min−1) |
|---|
| 8-Nitro-cGMP | Rp-8-Nitro-cGMPS |
|---|
| Cysteine | 0.255±0.014 | 0.27±0.011 |
| Glutathione | 0.105±0.004 | 0.11±0.005 |
8-Nitro-cGMP (20 µM) or Rp-8-nitro-cGMPS (20 µM) was reacted with low-molecular-weight thiols (10 mM) in 200 mM sodium phosphate buffer (pH 7.4) at 37°C.
8-Nitro-cGMP reacts with cysteine residues in proteins to induce posttranslational modification (protein S-guanylation). We studied whether Rp-8-nitro-cGMPS would induce a protein S-guanylation-like modification of PKG1α. Western blotting with the use of anti-S-guanylation antibody clearly suggested the occurrence of an S-guanylation-like modification of PKG1α after reaction of the enzyme with Rp-8-nitro-cGMPS, to a similar extent as that for the reaction between PKG1α and 8-nitro-cGMP (Fig. 4). If we take the electrophilicity data just described into consideration, Rp-8-nitro-cGMPS may bind to PKG1α followed by reacting with cysteine residues in the enzyme to induce an S-guanylation-like modification.
Inhibitory Activity of Rp-8-Nitro-cGMPS against PKG1αWe investigated the effects of Rp-8-nitro-cGMPS on the enzymatic activity of PKG1α by using recombinant enzyme with the natural activator cGMP. Rp-8-Bromo-cGMPS was used for comparison. Rp-8-Bromo-cGMPS inhibited PKG1α activity in a dose-dependent manner. Dixon plot analysis showed that Rp-8-bromo-cGMPS acted as a competitive inhibitor with a Ki value of 2.5 µM (Fig. 5A). This observed Ki value agreed with the reported value (3.7 µM).22) We found that Rp-8-nitro-cGMPS also inhibited the enzymatic activity of PKG1α in a competitive manner, with a Ki value of 22.5 µM (Fig. 5B). We thus suggest that Rp-8-nitro-cGMPS, like Rp-cGMPS derivatives, binds to the regulatory domain of PKG1α, but it does not cause an enzymatic conformational change for full activation of the enzyme.
Effects of Rp-8-Nitro-cGMPS on Vascular ResponsesWith vessels precontracted by phenylephrine, acetylcholine induced a concentration-dependent relaxation (Fig. 6A). During this vascular relaxation, NO derived from endothelial cells activates cGMP formation in smooth muscle cells and leads to PKG1α activation. 8-Bromo-cGMP is a cell-permeable analogue of cGMP and can induce vascular relaxation by direct activation of PKG1α (Fig. 6B). Rp-8-Bromo-cGMPS reduced vascular relaxation induced by both acetylcholine and 8-bromo-cGMP (Fig. 6). As an interesting finding, stronger inhibition of acetylcholine- and 8-bromo-cGMP-induced vascular relaxation was observed for Rp-8-nitro-cGMPS-treated vessels than for Rp-8-bromo-cGMPS-treated vessels (Fig. 6).
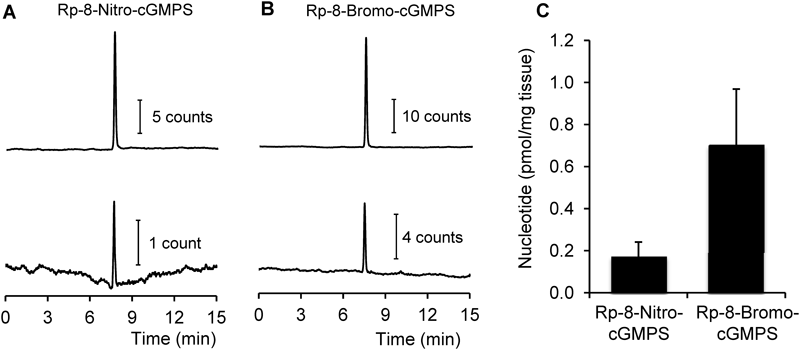
Tissue distribution of Rp-8-nitro-cGMPS in mouse aorta was quantified by means of LC-MS/MS. As Fig. 7A illustrates, a peak corresponding to Rp-8-nitro-cGMPS was clearly found in mouse aorta homogenates. Quantitative analysis revealed that tissue levels of Rp-8-nitro-cGMPS were approximately one quarter of those of Rp-8-bromo-cGMPS when mouse aorta rings were treated with the compounds at 100 µM for 1 h (Figs. 7B, C).
Inhibition of VASP Phosphorylation in C6 Cells by Rp-8-Nitro-cGMPSVASP is one of the founding members of a family of proline-rich proteins known as Eva/VASP proteins, and known as a substrate for PKG activity.20,21) Phosphorylation of VASP at Ser239 has been used to assess NO/cGMP/PKG signaling pathway activity.20,21) In this study, phosphorylation of VASP at Ser239 was clearly observed in rat glioma C6 cells by NO donor treatment in time- and dose-dependent manner (Fig. 8A). When cells were treated with Rp-8-nitro-cGMPS prior to NO donor stimulation, VASP phosphorylation was remarkably attenuated (Figs. 8B, C). This data suggests that Rp-8-nitro-cGMPS potentially inhibits PKG activity in C6 cells. Under the condition, Rp-8-bromo-cGMPS failed to inhibit PKG activity, suggesting that inhibitory action of Rp-8-nitro-cGMPS was stronger than that of Rp-8-bromo-cGMPS.
DISCUSSION
The structure of PKG1α consists of several functionally different domains1,3,23) (Fig. 8). The N-terminal domain with a coiled-coil structure promotes the formation of a PKG1α homodimer. The catalytic domain consists of ATP-binding site and substrate-binding site. In the resting state, the substrate-binding site in the catalytic domain is covered by an autoinhibitory site to limit the access of substrates. The binding of cGMP to the cGMP-binding sites located in the regulatory domain of PKG1α induces the conformational change necessary for full kinase activity (Fig. 8). Several types of PKG inhibitors have been developed to target different sites and domains in the enzyme.4,5) Cyclic nucleotide analogues such as Rp-cGMPS bind to the cGMP-binding sites without induction of a conformational change in the enzyme.6) K-Series and H-series inhibitors are low-molecular-weight inhibitors that bind to the ATP-binding site of PKG for enzyme inhibition.4) DT inhibitors are peptide inhibitors that target the substrate-binding region of the enzyme.4)
Monophosphorothioate analogues of cGMP (cGMPS) have dual effects on PKG1 activation that depend on the stereochemistry of the compounds.6) The Sp-diastereomer (Sp-cGMPS), which has an axial exocyclic sulfur atom in the cyclic phosphate moiety, binds to PKG1 and acts as an agonist.6) In contrast, the Rp-diastereomer (Rp-cGMPS), with an equatorial exocyclic sulfur atom, binds to the enzyme and acts as an antagonist.6) In this study, we synthesized Rp-8-nitro-cGMPS as the nitrated analogue of Rp-cGMPS and studied its effects on PKG1α activity and vascular responses. Similar to the reaction between 8-bromo-cGMP and sodium nitrite, as reported previously,7,9) the reaction of Rp-8-bromo-cGMPS with sodium nitrite resulted in the formation of Rp-8-nitro-cGMPS (Fig. 1). Rp-8-Bromo-cGMPS that remained in the reaction mixtures was removed by using semi-preparative RP-HPLC (Fig. 1, Supplementary Fig. S1).
As mentioned earlier, Rp-cGMPS binds to the same cGMP-binding sites in PKG1α but does not induce a conformational change in the enzyme, so it acts as a competitive inhibitor.6) We demonstrated in this study that Rp-8-nitro-cGMPS is indeed a competitive inhibitor of PKG1α (Fig. 5B). The Ki value of Rp-8-nitro-cGMP was 22.5 µM, which is larger than that for Rp-8-bromo-cGMPS (3.7 µM)22) and quite close to that reported for Rp-cGMPS (20 µM).6) This finding suggests that Rp-8-nitro-cGMPS has a binding affinity for cGMP-binding sites of PKG1α that is weaker than that of Rp-8-bromo-cGMPS but almost the same as that of Rp-cGMPS. We previously reported that 8-nitro-cGMP activated PKG1α with the apparent activation constant that was almost the same as that of native cGMP (110±5 nM for native cGMP and 119±3 nM for 8-nitro-cGMP).12) These data together indicate that nitration occurring at the C8 position may not affect the binding affinity of cyclic nucleotide derivatives for the cGMP-binding sites of PKG1α.
We found here that Rp-8-nitro-cGMPS inhibited vascular relaxation caused by both acetylcholine and 8-bromo-cGMP with more potency than did Rp-8-bromo-cGMPS (Fig. 6). As just discussed, the binding affinity of Rp-8-nitro-cGMPS to PKG1α is low compared with that of Rp-8-bromo-cGMPS (Fig. 5). The lipophilicity of PKG inhibitors has been thought to be an important determinant of the inhibitory potentials of PKG inhibitors in cell and tissue experiments.4,5) We previously determined the lipophilicity of 8-nitro-cGMP and 8-bromo-cGMP on the basis of octanol/water partition coefficient measurements, and we found 8-nitro-cGMP to be less lipophilic than 8-bromo-cGMP.12) Consistent with this result, we found that tissue levels of Rp-8-nitro-cGMPS were lower than those of Rp-8-bromo-cGMPS after mouse aortic rings were incubated with those inhibitors (Fig. 7). Therefore, the difference in binding affinity, lipophilicity, or tissue-penetrating ability of the inhibitors may not be the major factor for the greater inhibition of vascular relaxation by Rp-8-nitro-cGMPS compared with Rp-8-bromo-cGMPS. One possible explanation for such stronger inhibition of vascular relaxation by Rp-8-nitro-cGMPS is that it can cause covalent attachment of the Rp-cGMPS moiety to the enzyme via a protein S-guanylation-like modification (Fig. 4). This S-guanylation-like modification of PKG1α may result in persistent and irreversible enzyme inhibition. However, Rp-8-bromo-cGMPS may be in equilibrium in cells and tissues in both the enzyme-bound state and the free state, and hence the effect on PKG1α inhibition is reversible.4) This notion is supported by the finding in in vitro cell culture experiments that PKG inhibition was clearly observed after long term incubation (4 h) with Rp-8-nitro-cGMPS (Fig. 8).
As shown in Supplementary Fig. S3, PKG1α contains 11 Cys residues. Ten out of 11 Cys residues, except for Cys42, are conserved in PKG1β. On the other hand, 7 Cys residues are conserved in PKG2. S-Guanylation occurring at Cys195 is indispensable for persistent activation of PKG1α by 8-nitro-cGMP.12) It is thus hypothesized that Rp-8-nitro-cGMPS may inhibit PKG1β activity via S-guanylation-like modification, whereas effect of Rp-8-nitro-cGMPS on PKG2 may be limited because the enzyme lacks key Cys residue equivalent to Cys195 of PKG1α. Further study is needed to clarify the specificity of inhibitory action of Rp-8-nitro-cGMPS on PKG isotypes.
Although clinical applications of PKG inhibitors have not yet been demonstrated, PKG inhibition may be a potential target in the treatment of certain diseases such as bacterial diarrhea24) and cancers. In cancer cells, PKG1α signaling may suggest enhanced anti-apoptotic ability and thus chemoresistance of ovarian cancers to anticancer drugs.25) The PKG1α inhibitor DT-2 significantly increases spontaneous apoptosis in the human lung carcinoma cell lines H460 and A549.26) Inhibition of PKG1α signaling by this same DT-2 inhibitor dramatically enhanced pro-apoptotic effects of the chemotherapeutic agent cisplatin in those cancer cell lines.26) PKG inhibitors may be used to prevent migration and invasion of colon cancer cells. In scratch wound and modified Boyden chamber assays with colorectal carcinoma cell lines, the PKG inhibitor KT5823 effectively halted cell migration induced by NO donor treatment.27) In contrast to beneficial effects of PKG inhibitors in cancer treatment, one report suggested a pro-apoptotic role for PKG in estrogen receptor-positive (MCF-7) and estrogen receptor-negative (MDA-MB-468) breast cancer cell lines.28) Therefore, to make use of PKG inhibitors in cancer treatment, a specific classification of the malignancy and precise details of its response to treatment are critical.
In addition to PKG1α, a number of proteins have been identified as endogenous targets of protein S-guanylation. Biological consequences regulated by protein S-guanylation vary dependent on target proteins and sites to be modified.11,29,30) Kelch-like ECH-associated protein 1 (Keap1) was the first endogenous target of protein S-guanylation identified in cells.7,8) Keap1 binds to the transcription factor nuclear factor (erythroid-derived 2)-like 2 (Nrf2) and acts as a negative regulator of Nrf2.31) S-Guanylation at Cys434 of Keap1 may facilitate dissociation of the protein from Nrf2, which would lead to activation of Nrf2-dependent gene expression.31) Other targets of protein S-guanylation identified so far include mitochondrial heat shock protein-60,32) small GTPase H-Ras,9) SNAP25 in the SNARE complex,33) and tau protein.34) We found that 8-nitro-cGMP causes protein S-guanylation of H-Ras in the mouse heart during myocardial infarction and in rat cardiac fibroblasts in culture.9) In vitro analyses clearly demonstrated that 8-nitro-cGMP treatment markedly activates the H-Ras-dependent signaling pathway including activation of extracellular signal-regulated kinase (ERK), p38 mitogen-activated protein kinase (p38 MAPK), and p53 in rat cardiomyocytes.9) The rat cardiac fibroblast culture model revealed that S-guanylation of H-Ras at Cys186 promotes a change in H-Ras localization from raft to nonraft regions, which leads to full enzyme activation by inhibiting palmitoylation at the same cysteine residue. Protein S-guanylation influences protein complex formation as shown by the inhibition by S-guanylation of the aggregation of tau protein.34) 8-Nitro-cGMP treatment significantly reduces the amount of sarkosyl-insoluble tau protein in tau-expressing Neuro2A cells.34) NO-linked chemical modification of the cysteine residues in tau protein blocks aggregation of tau protein, thus increasing brain 8-nitro-cGMP levels, which may become a therapeutic strategy in Alzheimer’s disease. The occurrence of protein S-guanylation in prokaryotes was also recently identified in the Gram-negative bacterium Escherichia coli.35) A number of proteins, including chaperones, ribosomal proteins, and metabolic and redox regulation enzymes in E. coli, are susceptible to S-guanylation. Because Rp-8-nitro-cGMPS possesses electrophilicity that is almost the same as that of 8-nitro-cGMP (Fig. 3), additional study is needed to clarify whether Rp-8-nitro-cGMPS treatment modulates cellular responses regulated by the above-mentioned proteins via a protein S-guanylation-like modification.
In conclusion, this study demonstrated that Rp-8-nitro-cGMPS serves as a potent inhibitor of PKG1α by competing with binding of cGMP to the regulatory domain of the enzyme (Fig. 9). Covalent attachment of the Rp-cGMPS moiety to the regulatory domain of the enzyme via a protein S-guanylation-like modification suggests potent inhibition of vascular relaxation in Rp-8-nitro-cGMPS treatment. These findings warrant continued investigation to establish applications of Rp-8-nitro-cGMPS as a biochemical tool in signal transduction and as a potent inhibitor for therapeutic purposes.
Acknowledgments
We thank J. B. Gandy for her editing of the manuscript. This work was supported in part by Grants-in-Aid for Scientific Research [(A), (B), Innovative Areas (Research in a Proposed Area), and Research Activity Start-up] from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, to K.O. (15H06509), T.A. (26111008, 26111001, 15K21759, 25253020, 16K15208), and T.S. (15H03115).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev., 62, 525–563 (2010).
- 2) Smolenski A, Burkhardt AM, Eigenthaler M, Butt E, Gambaryan S, Lohmann SM, Walter U. Functional analysis of cGMP-dependent protein kinases I and II as mediators of NO/cGMP effects. Naunyn Schmiedebergs Arch. Pharmacol., 358, 134–139 (1998).
- 3) Hofmann F, Bernhard D, Lukowski R, Weinmeister P. cGMP regulated protein kinases (cGK). Handb. Exp. Pharmacol., 191, 137–162 (2009).
- 4) Wolfertstetter S, Huettner JP, Schlossmann J. cGMP-dependent protein kinase inhibitors in health and disease. Pharmaceuticals, 6, 269–286 (2013).
- 5) Schwede F, Maronde E, Genieser H, Jastorff B. Cyclic nucleotide analogs as biochemical tools and prospective drugs. Pharmacol. Ther., 87, 199–226 (2000).
- 6) Butt E, van Bemmelen M, Fischer L, Walter U, Jastorff B. Inhibition of cGMP-dependent protein kinase by (Rp)-guanosine 3′,5′-monophosphorothioates. FEBS Lett., 263, 47–50 (1990).
- 7) Sawa T, Zaki MH, Okamoto T, Akuta T, Tokutomi Y, Kim-Mitsuyama S, Ihara H, Kobayashi A, Yamamoto M, Fujii S, Arimoto H, Akaike T. Protein S-guanylation by the biological signal 8-nitroguanosine 3′,5′-cyclic monophosphate. Nat. Chem. Biol., 3, 727–735 (2007).
- 8) Fujii S, Sawa T, Ihara H, Tong KI, Ida T, Okamoto T, Ahtesham AK, Ishima Y, Motohashi H, Yamamoto M, Akaike T. The critical role of nitric oxide signaling, via protein S-guanylation and nitrated cyclic GMP, in the antioxidant adaptive response. J. Biol. Chem., 285, 23970–23984 (2010).
- 9) Nishida M, Sawa T, Kitajima N, Ono K, Inoue H, Ihara H, Motohashi H, Yamamoto M, Suematsu M, Kurose H, van der Vliet A, Freeman BA, Shibata T, Uchida K, Kumagai Y, Akaike T. Hydrogen sulfide anion regulates redox signaling via electrophile sulfhydration. Nat. Chem. Biol., 8, 714–724 (2012).
- 10) Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. U.S.A., 111, 7606–7611 (2014).
- 11) Sawa T, Ihara H, Ida T, Fujii S, Nishida M, Akaike T. Formation, signaling functions, and metabolisms of nitrated cyclic nucleotide. Nitric Oxide, 34, 10–18 (2013).
- 12) Akashi S, Ahmed KA, Sawa T, Ono K, Tsutsuki H, Burgoyne JR, Ida T, Horio E, Prysyazhna O, Oike Y, Rahaman MM, Eaton P, Fujii S, Akaike T. Persistent activation of cGMP-dependent protein kinase by a nitrated cyclic nucleotide via site specific protein S-guanylation. Biochemistry, 55, 751–761 (2016).
- 13) Zaki MH, Fujii S, Okamoto T, Islam S, Khan S, Ahmed KA, Sawa T, Akaike T. Cytoprotective function of heme oxygenase 1 induced by a nitrated cyclic nucleotide formed during murine salmonellosis. J. Immunol., 182, 3746–3756 (2009).
- 14) Ahmed KA, Sawa T, Ihara H, Kasamatsu S, Yoshitake J, Rahaman MM, Okamoto T, Fujii S, Akaike T. Regulation by mitochondrial superoxide and NADPH oxidase of cellular formation of nitrated cyclic GMP: potential implications for ROS signalling. Biochem. J., 441, 719–730 (2012).
- 15) Kurauchi Y, Hisatsune A, Isohama Y, Sawa T, Akaike T, Shudo K, Katsuki H. Midbrain dopaminergic neurons utilize nitric oxide/cyclic GMP signaling to recruit ERK that links retinoic acid receptor stimulation to up-regulation of BDNF. J. Neurochem., 116, 323–333 (2011).
- 16) Ihara H, Ahtesham AK, Ida T, Kasamatsu S, Kunieda K, Okamoto T, Sawa T, Akaike T. Methodological proof of immunochemistry for specific identification of 8-nitroguanosine 3′,5′-cyclic monophosphate formed in glia cells. Nitric Oxide, 25, 169–175 (2011).
- 17) Kasamatsu S, Watanabe Y, Sawa T, Akaike T, Ihara H. Redox signal regulation via nNOS phosphorylation at Ser847 in PC12 cells and rat cerebellar granule neurons. Biochem. J., 459, 251–263 (2014).
- 18) Joudoi T, Shichiri Y, Kamizono N, Akaike T, Sawa T, Yoshitake J, Yamada N, Iwai S. Nitrated cyclic GMP modulates guard cell signaling in Arabidopsis. Plant Cell, 25, 558–571 (2013).
- 19) Honda K, Yamada N, Yoshida R, Ihara H, Sawa T, Akaike T, Iwai S. 8-Mercapto-cyclic GMP mediates hydrogen sulfide-induced stomatal closure in Arabidopsis. Plant Cell Physiol., 56, 1481–1489 (2015).
- 20) Cook ALM, Haynes JM. Phosphorylation of the PKG substrate, vasodilator-stimulated phosphoprotein (VASP), in human cultured prosatic stromal cells. Nitric Oxide, 16, 10–17 (2007).
- 21) Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol., 19, 541–564 (2003).
- 22) Kawada T, Toyosato A, Islam MO, Yoshida Y, Imai S. cGMP-kinase mediates cGMP- and cAMP-induced Ca2+ desensitization of skinned rat artery. Eur. J. Pharmacol., 323, 75–82 (1997).
- 23) Alverdi V, Mazon H, Versluis C, Hemrika W, Esposito G, van den Heuvel R, Scholten A, Heck AJ. cGMP-binding prepares PKG for substrate binding by disclosing the C-terminal domain. J. Mol. Biol., 375, 1380–1393 (2008).
- 24) Pfeifer A, Aszodi A, Seidler U, Ruth P, Hofmann F, Fassler R. Intestinal secretory defects and dwarfism in mice lacking cGMP-dependent protein kinase II. Science, 274, 2082–2086 (1996).
- 25) Leung EL, Wong JC, Johlfs MG, Tsang BK, Fiscus RR. Protein kinase G type Iα activity in human ovarian cancer cells significantly contributes to enhanced Src activation and DNA synthesis/cell proliferation. Mol. Cancer Res., 8, 578–591 (2010).
- 26) Wong JC, Bathina M, Fiscus RR. Cyclic GMP/protein kinase G type-Iα (PKG-Iα) signaling pathway promotes CREB phosphorylation and maintains higher c-IAP1, livin, survivin, and Mcl-1 expression and the inhibition of PKG-Iα kinase activity synergizes with cisplatin in non-small cell lung cancer cells. J. Cell. Biochem., 113, 3587–3598 (2012).
- 27) Babykutty S, Suboj P, Srinivas P, Nair AS, Chandramohan K, Gopala S. Insidious role of nitric oxide in migration/invasion of colon cancer cells by upregulating MMP-2/9 via activation of cGMP-PKG-ERK signaling pathways. Clin. Exp. Metastasis, 29, 471–492 (2012).
- 28) Fallahian F, Karami-Tehrani F, Salami S, Aghaei M. Cyclic GMP induced apoptosis via protein kinase G in oestrogen receptor-positive and -negative breast cancer cell lines. FEBS J., 278, 3360–3369 (2011).
- 29) Sawa T, Arimoto H, Akaike T. Regulation of redox signaling involving chemical conjugation of protein thiols by nitric oxide and electrophiles. Bioconjug. Chem., 21, 1121–1129 (2010).
- 30) Fujii S, Sawa T, Nishida M, Ihara H, Ida T, Motohashi H, Akaike T. Redox signaling regulated by an electrophilic cyclic nucleotide and reactive cysteine persulfides. Arch. Biochem. Biophys., 595, 140–146 (2016).
- 31) Taguchi K, Motohashi H, Yamamoto M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells, 16, 123–140 (2011).
- 32) Rahaman MM, Sawa T, Ahtesham AK, Khan S, Inoue H, Irie A, Fujii S, Akaike T. S-Guanylation proteomics for redox-based mitochondrial signaling. Antioxid. Redox Signal., 20, 295–307 (2014).
- 33) Kunieda K, Tsutsuki H, Ida T, Kishimoto Y, Kasamatsu S, Sawa T, Goshima N, Itakura M, Takahashi M, Akaike T, Ihara H. 8-Nitro-cGMP enhances SNARE complex formation through S-guanylation of Cys90 in SNAP25. ACS Chem. Neurosci., 6, 1715–1725 (2015).
- 34) Yoshitake J, Soeda Y, Ida T, Sumioka A, Yoshikawa M, Matsushita K, Akaike T, Takashima A. Modification of Tau by 8-nitroguanosine 3′,5′-cyclic monophosphate (8-nitro-cGMP): effects of nitric oxide-linked chemical modification on tau aggregation. J. Biol. Chem., 291, 22714–22720 (2016).
- 35) Tsutsuki H, Jung M, Zhang T, Ono K, Ida T, Kunieda K, Ihara H, Akaike T, Sawa T. Endogenous occurrence of protein S-guanylation in Escherichia coli: target identification and genetic regulation. Biochem. Biophys. Res. Commun., 478, 7–11 (2016).