Current Topics: Reviews
The Role of mPGES-1 in Inflammatory Brain Diseases
2017 Volume 40 Issue 5 Pages 557-563
Details
2017 Volume 40 Issue 5 Pages 557-563
Prostaglandin E2 (PGE2) has been thought to be an important mediator of inflammation in peripheral tissues, but recent studies clearly show the involvement of PGE2 in inflammatory brain diseases. In some animal models of brain disease, the genetic disruption and chemical inhibition of cyclooxygenase (COX)-2 resulted in the reduction of PGE2 and amelioration of symptoms, and it had been thought that PGE2 produced by COX-2 may be involved in the progression of injuries. However, COX-2 produces not only PGE2, but also some other prostanoids, and thus the protective effects of COX-2 inhibition, as well as severe side effects, may be caused by the inhibition of prostanoids other than PGE2. Therefore, to elucidate the role of PGE2, studies of microsomal prostaglandin E synthase-1 (mPGES-1), an inducible terminal enzyme for PGE2 synthesis, have recently been an active area of research. Studies from mPGES-1 deficient mice provide compelling evidence for its role in a variety of inflammatory brain diseases, such as ischemic stroke, Alzheimer’s disease and epilepsy, and clues for developing new therapeutic treatments for brain diseases by targeting mPGES-1. Considering that COX inhibitors may non-selectively suppress the production of many types of prostanoids that are essential for normal physiological functioning of the brain and peripheral tissues, as well as induce gastro-intestinal, renal and cardiovascular complications, mPGES-1 inhibitors are expected to be injury-selective and have fewer side-effects when treating human brain diseases. Thus, this paper focuses on recent studies that have demonstrated the involvement of mPGES-1 in pathological brain diseases.
Prostaglandin E2 (PGE2) is one of the most common prostaglandins in the brain and contributes to physiological and pathophysiological brain functions. Under physiological conditions, PGE2 is known to modulate neuronal excitation and plasticity, thereby contributing to learning and memory.1,2) PGE2 also contributes to neuronal proliferation and differentiation and dendritic spine formation.3,4) PGE2 is believed to be an important mediator of inflammation in peripheral tissues, but recent studies have clearly shown the involvement of PGE2 in brain diseases. Under pathological conditions, excess PGE2 is known to be produced in lesion sites in the brain and contributes to the progression of symptoms, while in some cases, it rather ameliorates the progression.5,6)
PGE2 is derived from membrane phospholipids through three sequential enzymatic processes; phospholipase A2 (PLA2) which releases arachidonic acid (AA) from membrane phospholipids, cyclooxygenase (COX) which converts AA to PGH2, and prostaglandin E synthase (PGES) which isomerize PGH2 to PGE2 (Fig. 1). After synthesis, PGE2 can efflux by simple diffusion or by a PGE2 efflux transporter, such as MRP4, and activate four G-protein-coupled receptors, EP1–EP4, with quite different signaling cascades (Fig. 1). The EP1 and EP3 receptors appear to be essential for the neurotoxicity mediated by PGE2, while the EP2 receptor appears to be protective, determining the scope of acute neuronal injury.6–8)

Arachidonic acid is metabolized by cyclooxygenase (COX: COX-1 is the stable form and COX-2 is the inducible form) to the unstable endoperoxide PGH2, and then metabolized by prostaglandin E synthase (PGES: cPGES and mPGES-2 are the stable forms and mPGES-1 is the inducible form) to PGE2. PGH2 is also metabolized to PGD2, PGI2 and TXA2 by PGDS, PGIS and TXS, respectively. PGE2 activates four G-protein-coupled receptors, EP1–EP4, with quite different signaling cascades to induce cellular responses.
Among the COX isoforms, COX-2 is the inducible form, however, it has been immunohistochemically detected in neurons in the normal brain.9) A large number of studies have demonstrated a neurotoxic role for COX-2 in a broad spectrum of inflammatory brain disease models, such as cerebral ischemia, neurodegenerative diseases, and traumatic brain injury.5) In these brain disease models, since the genetic disruption and chemical inhibition of COX-2 resulted in the reduction of PGE2 and amelioration of symptoms, it had been thought that PGE2 produced by COX-2 may be involved in the progression of injuries. However, COX-2 produces not only PGE2, but also PGI2, PGD2, PGF2α and thromboxane (TX) A2 (Fig. 1), thus, inhibition of prostanoids other than PGE2 may contribute to the neuroprotective effects of genetic disruption or pharmacological inhibition of COX-2. Therefore, to elucidate the role of PGE2, studies on PGES, the terminal enzyme for PGE2 synthesis, have recently become an active area of research. In this paper, I focus on the role of mPGES-1, an inducible enzyme for PGE2 synthesis, in pathological brain conditions.
mPGES-1 is a microsomal, glutathione-dependent and inducible enzyme, and a member of the MAPEG (membrane-associated proteins involved in eicosanoid and glutathione metabolism) protein superfamily, which was cloned in 1999.10) Among three major isoforms of PGES, cytosolic PGES (cPGES), mPGES-1 and mPGES-2, only mPGES-1 is an inducible enzyme, which is upregulated by proinflammatory stimuli and in various models of inflammatory brain diseases.11) Recently, not only mPGES-1, but also cPGES and mPGES-2, have been reported to be upregulated in certain human brain diseases, such as glioma and Alzheimer’s disease (AD).12,13)
In many pathological conditions, mPGES-1 is co-induced with COX-2 in the same cells in the brain, such as microglia, endothelial cells and neurons.14,15) Indeed, mPGES-1 was shown to functionally couple to COX-2.16) However, in some cases, mPGES-1 is co-induced with COX-1 to mediate PGE2 elevation in the brain.17) In spite of co-induction of mPGES-1 and COX-2 in the same cells, evidence for distinct signaling pathways leading to mPGES-1 and COX-2 induction was observed in a study where phosphatidylinositol 3-kinase inhibition effectively uncoupled co-regulation of these two enzymes in rat microglia treated with lipopolysaccharide (LPS).18) Indeed, a difference in the time courses of the induction between mPGES-1 and COX-2 protein have been reported in several conditions, including an animal model of ischemic stroke14) and LPS-stimulated microglia,19) showing persistent induction of mPGES-1 and transient induction of COX-2. These results suggest a difference in the mechanisms underlying the inductions of mPGES-1 and COX-2, that are thought to be controlled mainly by transcription factors, Egr-1 and NF-κB, respectively.20)
The profile of mPGES-1 deficient mice strongly supports the idea that mPGES-1 plays an important role in the inflammation in animal models of pain, arthritis and pyrexia.11) Recently, the involvement of mPGES-1 in inflammatory brain diseases has also been reported by showing induction of mPGES-1 in brain lesion sites of postmortem patients and/or animal models of inflammatory brain diseases and that genetic disruption of mPGES-1 ameliorates the symptoms of these diseases as described below (Table 1). Therefore, mPGES-1 has been thought of as a potential novel target for inflammatory brain diseases. Additional information about mPGES-1, its role in peripheral inflammatory diseases, and potential as a therapeutic target can be found in a recent review.11,21)
| Diseases | Cells inducing mPGES-1 expression | Functions | References |
|---|---|---|---|
| Brain ischemia | Rat (MCAO model) | Infarction | 14, 28) |
| Microglia (core) | Edema | ||
| Endothelial cells (core) | Neuronal apoptosis | ||
| Neurons (peri) | Neurological deficit | ||
| Alzheimer’s disease | Human (AD patients) | Neuronal apoptosis | 25, 35, 36) |
| Pyramidal neurons | Aβ accumulation | ||
| Dystrophic neurites surrounding senile Aβ | Glial activation | ||
| Tg2576 mouse (AD model) | Learning and memory | ||
| Astrocytes | |||
| Mouse (Aβ peptide stimulation in vitro) | |||
| Neurons | |||
| Parkinson’s disease | Rat (Intranigral LPS injection model) | Microglial activation | 19) |
| Microglia | |||
| Epilepsy | Rat/mouse (KA injection model) | Glutamate release (astrocytes) | 41, 42) |
| Endothelial cells | Neuronal damage | ||
| Brain cancer | Human (GBM) | Glioma proliferation | 13, 45, 46, 47, 48) |
| Glioma | Endothelial cells in the peritumoral tissue | Tumor growth | |
| Neuroblastoma | Glioblastoma | Immunosuppression | |
| Human (Neuroblastoma) | Glioma apoptosis | ||
| Cancer-associated fibroblasts | |||
| Mouse (in vitro) | |||
| Macrophages, microglia |
In experimental animal models and patients with brain diseases, mPGES-1 has been shown to be upregulated in the lesion site of the brain.14,15) In most of these reports, the immunostaining for mPGES-1 is rarely detectable in the brain of control groups and outside of the lesion site of the disease model groups, while significant staining is often observed in the lesion site of the brain. Therefore, like in many other tissues, basal mPGES-1 expression in the brain seems to be very low. In the rat brain, mPGES-1 was reported to be constitutively present in postsynaptic dendrites.22) It is also reported that normal adult rat brain cortex constitutively expressed neuronal perinuclear mPGES-1 and COX-2.23) Recently, the distribution of mPGES-1 in the mouse brain has been reported, showing that mPGES-1 was constitutively expressed in endothelial cells, capillary-associated pericytes, astrocytes, leptomeninges, and the choroid plexus of mouse brain.24) Regional differences were shown with prominent labeling in autonomic relay structures such as the area postrema, subfornical organ, paraventricular hypothalamic nucleus, arcuate nucleus, and preoptic area. Actually, we observed that the PGE2 level in the cerebral cortex of wild-type mice was more than twice that of mPGES-1 deficient mice, showing the contribution of mPGES-1 to basal PGE2 production in the mouse brain (Fig. 2C, SHAM-operated control group). Immunostaining of the human brain also showed that mPGES-1 was normally expressed constitutively in neurons, microglia, astrocytes, and endothelial cells.25) Therefore, even though there are no apparent phenotypic differences in brain structure or behavior between mPGES-1 deficient mice and wild-type mice,14) the role of constitutively expressed mPGES-1 in the human brain should be elucidated to avoid any possible side-effects of mPGES-1 inhibitors in future clinical use.

A. Immunostaining for mPGES-1 and COX-2 and Nissl staining of a coronal rat brain slice 24 h after ischemia. Representative data from 6 animals are presented. B. Representative TTC-stained coronal sections of mPGES-1 deficient (−/−) and wild-type (+/+) mice. C. The production of PGE2 in the ipsilateral (i) or contralateral (c) cortex of mPGES-1 deficient (−/−) and wild-type (+/+) mice 24 h after MCA occlusion (MCAO) and sham operation (SHAM). n=8 animals per group, ** p<0.01 vs. another sample. Scale bar, 5 mm. Modified from ref. 14. “Copyright (2006) National Academy of Sciences.”
Stroke remains one of the major causes of death and neurological disability throughout the world. Treatments based on thrombolysis and restoration of blood flow are effective only during the first few hours after the onset of an ischemic stroke. At the later phase, brain inflammation was reported to be a major factor in the progression of the neuronal damage.26) Genetic inhibition of COX-2, but not COX-1, and pharmacological inhibition of COX have exhibited beneficial effects in animal models,27,28) while neuronal overexpression of COX-2 increases cerebral infarction.29) Thus, it has been thought that PGE2, a major end product of COX, may play a pathogenic role in ischemic stroke.
Recently, we reported on the role of mPGES-1 in ischemic brain injury using a rodent middle cerebral artery occlusion-reperfusion (MCAO) model.14) Both mPGES-1 and COX-2, but not cPGES, mPGES-2 and COX-1, were induced (Fig. 2A) and co-localized in neurons in the peri-infarct region, and in microglia and endothelial cells in the ischemic core region of the cerebral cortex after transient focal ischemia. Similar to the results from other models, COX-2 protein expression was more transiently expressed and earlier in time, whereas mPGES-1 expression persisted with maximal expression between days 1 and 3 before returning to baseline at day 7, and PGE2 biosynthesis peaked on 1 d after ischemia. In mPGES-1 deficient mice in which postischemic PGE2 production in the cortex was completely absent (Fig. 2C), we found that ischemic injuries such as infarction (Fig. 2B), edema, and increase in terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick-end labeling (TUNEL)-positive apoptotic cells were significantly reduced compared to those in wild-type mice. Disruption of mPGES-1 also ameliorated the behavioral symptoms, such as neurological dysfunction and reduction in locomotor activity, observed after ischemia. There were no significant differences between mPGES-1 deficient and wild-type mice in mean arterial pressure, pH, pCO2, or pO2 levels, or the changes in cerebral blood flow before, during, or after middle cerebral artery occlusion. The ischemic symptoms in mPGES-1 deficient mice were less severe than those in wild-type mice and the intracerebroventricular injection of an appropriate amount of PGE2 into deficient mice reversed the ischemic symptoms to almost the same severity as wild-type mice, suggesting the involvement of mPGES-1 in the exaggeration of ischemic injury through PGE2 production. The induction and involvement of mPGES-1 in neurotoxicity were also observed in an in vitro excitotoxicity model.28) Exposure to glutamate increased the expression of mPGES-1 and production of PGE2 in rat and mouse hippocampal slices. NS-398, an inhibitor of COX-2, ameliorated the glutamate-induced excitotoxicity in wild-type slices, but not in mPGES-1 deficient slices, which showed less toxicity than wild-type slices. Similar to the results from an in vitro model, NS-398 reduced not only ischemic PGE2 production, but also ischemic injuries in wild-type mice, but not in mPGES-1 deficient mice, which showed ameliorated symptoms in a focal ischemia model. The combination of a COX-2 inhibitor and mPGES-1 deficient mice clearly shows mPGES-1 is needed for COX-2 neurotoxicity observed after brain ischemia. Taken together, our observations indicate a critical role for mPGES-1 in ischemic brain injury. Because intraperitoneal injection of the EP3 antagonist ONO-AE3-240 ameliorated infarction, edema, apoptotic cell death and neurological dysfunction in wild-type mice, but not in mPGES-1 deficient mice, mPGES-1 toxicity is thought to be mediated through EP3 receptors.7) Indeed, EP3 deficient mice showed similar amelioration in ischemic symptoms as EP3 antagonist and mPGES-1 deficient mice.30) Our observations suggest that mPGES-1 may be a critical determinant of postischemic neurological dysfunction and a valuable therapeutic target for treatment of human stroke.
AD is the most common neurodegenerative disorder and form of dementia among the elderly, where dementia symptoms gradually worsen over a number of years. Two abnormal structures called senile plaques (SPs), which have abundant deposits of β-amyloid peptide (Aβ) fibrils, and neurofibrillary tangles, which consist of abnormal tau protein filaments, are prime suspects in the damaging and killing of neurons. Inflammatory reactions, such as glial activations and production of proinflammatory cytokines around SPs have been demonstrated to accompany the neurodegeneration in AD.31) Although epidemiological studies have shown that nonsteroidal anti-inflammatory drugs (NSAIDs) which inhibit COX activity can reduce the risk of developing AD,32) clinical trials of NSAIDs in AD patients have unfortunately not been very fruitful. The protective effects of NSAIDs have been also shown in a transgenic mouse model of AD demonstrating that NSAIDs reduced amyloid deposition and microglial activation.33) Furthermore, PGE2 has been found to be elevated in cerebrospinal fluid early in AD.34) Thus, it has been thought that PGE2 may play a pathogenic role in AD.
The induction of mPGES-1 protein in the brain of AD patients has been shown in both sporadic and familial AD.25) mPGES-1 was shown to be normally expressed constitutively in human neurons, microglia, and endothelial cells, but was up-regulated in pyramidal neurons of AD. Another study showed the induction of mPGES-1 in dystrophic neurites surrounding senile Aβ plaques in the brain of human AD patients, whereas it was upregulated in astrocytes around the Aβ plaques in the Tg2576 mice, a transgenic AD mouse model.35) These differences in mPGES-1 inducing cells between AD patients and AD model mice might be due to differences in AD stages; autopsied end-stage AD brain tissues, in which severe neuronal cell death was induced, while severe neuronal cell death is not found in Tg2576 mice.
The involvement of mPGES-1 in AD pathology was examined by using primary neurons obtained from mPGES-1 deficient mice, which exhibit an absence of PGE2 production and less apoptosis after stimulation with Aβ fragment 31–35 (Aβ31–35).36) The combined treatment of Aβ31–35 and PGE2 induced apoptosis in mPGES-1 deficient neurons to the similar level as Aβ31–35-treated wild-type neurons. Furthermore, in an in vivo model of AD, genetic disruption of mPGES-1 in Tg2576 mice has been demonstrated to reduce the accumulation of microglia around SPs and attenuated learning impairments determined by the Morris water maze test.35) Thus, mPGES-1 is induced in the AD brain and plays a role in AD pathology, suggesting that blockage of mPGES-1 could form the basis for a novel therapeutic strategy for patients with AD.
Parkinson’s disease (PD) is the second most common age-related neurodegenerative disorder, after AD. It is characterized clinically by parkinsonism (resting tremor, bradykinesia, rigidity, and postural instability) and pathologically by the loss of dopaminergic neurons in the substantia nigra (SN) in association with the presence of abnormal protein deposits in the cytoplasm of neurons called Lewy bodies. Although there are many effective treatments available now, which mostly address motor symptoms, no known curative therapy exists. Recent extensive investigations including epidemiologic, animal, human, and therapeutic studies have revealed that neuroinflammation plays a key role in the initiation and progression of PD.31) PGE2, one of the most likely candidates for propagation of the inflammation, is known to accumulate in the SN of postmortem PD patients and animal models of PD induced by the neurotoxins 1-methyl-4-phenyl-1,2,4,6-tetrahydropyridine (MPTP) or 6-hydroxydopamine (6-OHDA).5) COX-2 has been demonstrated to be up-regulated in dopaminergic neurons and microglia of PD specimens and neurotoxin-induced animal models of PD. The genetic disruption and chemical inhibition of COX-2 have been shown to ameliorate dopaminergic neuronal death in neurotoxin-induced animal models of PD, suggesting that the PGE2 accumulated through COX-2 induction mediates the toxic effects in the brain.37)
There are still no reports demonstrating mPGES-1 induction in the brain of PD patients and neurotoxin-induced animal models of PD. The single intra-nigral injection of LPS has been shown to induce a strong macrophage/microglial reaction in the SN and cause neuronal damage in the dopaminergic neurons, but not in the GABAergic or serotoninergic neurons of the SN.38) Unlike the direct death of dopaminergic neurons caused by neurotoxins such as MPTP or 6-OHDA, intranigral injection of LPS seems to cause indirect death due to inflammatory reaction and is thought to be an interesting model for studying the selective effects of inflammatory reaction on the dopaminergic system and also potentially useful for studying PD. Using the rat intranigral LPS injection model, we have reported the induction of mPGES-1 specifically in the activated ameboid microglia in SN.19) We are now investigating the role of mPGES-1 using a neurotoxin-induced animal model of PD, and our preliminary data suggest the important role of mPGES-1 in dopaminergic neurodegeneration. Thus, mPGES-1 could be a novel therapeutic target for treatment of PD.
Epilepsy is the third most common chronic brain disorder, and is characterized by an abnormal excessive or synchronous neuronal activity that generates seizures. Several studies using an experimental animal model of epilepsy revealed that the effects of brain inflammation contribute to the generation of individual seizures and cell death, which, in turn, activates further inflammation, thereby establishing a vicious circle of events that contributes to the development of epilepsy.39)
In animal models of temporal lobe epilepsy induced by intrahippocampal or intraperitoneal injection of kainic acid (KA), an agonist of non-N-methyl-D-aspartate (non-NMDA) ionotropic glutamate receptors, COX-2 expression was shown to be increased immediately in hippocampal neurons and gradually in nonneuronal cells, such as endothelia and astrocytes, after induction of seizures.40) The PGE2 level was also increased in the hippocampus after KA-induced seizures. Late-induced COX-2 produces a large amount of PGE2 that may facilitate neuronal loss elicited by KA, because a COX-2 inhibitor relieved this neuronal damage. On the contrary, the neuroprotective effects of PGE2 have also been reported, thus, the effects of COX-2 inhibitors on the symptoms of epilepsy are still controversial.5,39)
The induction of mPGES-1 by KA has been recently reported in venous endothelial cells, and its deficiency significantly decreased not only hippocampal PGE2 production, but also hippocampal neuronal damage.41) The KA-induced elevation in astrocytic Ca2+ levels and release of glutamate in mPGES-1 deficient hippocampal slices were less than in wild-type slices, suggesting that PGE2 produced by endothelial mPGES-1 enhances astrocytic Ca2+ levels and increases Ca2+-dependent glutamate release, thereby aggravating neuronal injury.42) This novel endothelium–astrocyte–neuron signaling pathway may be crucial for neuronal damage after repetitive seizures, and hence could be a new target for drug development.
Malignant brain tumors including gliomas and neuroblastomas are among the most lethal of human tumors. Despite conventional treatments consisting of surgery, radiation, and chemotherapy, the prognosis of these tumors is extremely poor. Recent evidence suggests a crucial role for inflammation in brain tumors, showing that an inflammatory response promotes the survival of glioma cells and suppresses adaptive immune responses. Elevated levels of mPGES-1, COX-2 and PGE2 have been found in several different cancers, including colon cancer, non-small cell lung cancer, and prostate cancer, and have been shown to control the proliferation, survival, invasiveness and angiogenic potential of tumor cells.21) In the case of brain cancer, the overexpression of COX-2 in human gliomas was linked to increased aggressiveness and poor prognosis,43) and the involvement of COX-2 in glioma progression has been also reported in experimental models of glioma.44)
In human glioma, overexpression of mPGES-1, as well as mPGES-2 and cPGES, was observed in both low- and high-grade tumors.13) The analysis showed no correlation between tumor grade and PGES staining of tumor cells or vascular endothelium, except in oligodendrogliomas where moderate correlation could be found between tumor grade and tumor cell staining with mPGES-1 and cPGES. An increased expression of mPGES-1 in the tissues of patients of brain cancer has also been reported from another group in a subset of human glioblastoma multiforme (GBM) tumors, the most common form of adult brain cancer, astrocytoma and high-risk neuroblastoma.45–47) An activated COX/mPGES-1/PGE2 pathway has been identified in 11q-deleted neuroblastoma with high expression of mPGES-1, and elevated levels of PGE2 as compared with low-risk tumors. Analysis of expression cohorts revealed a worse outcome for high-risk patients (INSS stage 4) with high mPGES-1 expression.47) In accordance with the anti-apoptotic properties of COX-2, which is known to play an important role in tumor development, in an in vitro study using the human astroglioma cell line U-87MG, PGE2 produced by overexpression of mPGES-1 promoted glioma proliferation through activation of type 2 protein kinase A.46) COX inhibition in an in vivo model of 11q-deleted neuroblastoma also demonstrated that PGE2 produced by mPGES-1 resulted in promotion of tumor growth.47) On the other hand, intracellular PGE2 produced by mPGES-1 has been shown to promote apoptosis by activation of Bax through its physical interaction with PGE2.45) Thus, the balance between extracellular and intracellular PGE2 may be important in the control of apoptosis in glioma. An in vitro study using co-cultures of glioma cells with macrophages or microglia revealed that mPGES-1 was induced in macrophages and microglia by glioma-derived soluble factors and that PGE2 produced by mPGES-1 induced an immunosuppressive state in the central nervous system.48) Therefore, the manipulation of microglial PGE2 production provides an important strategy with which to induce an effective antiglioma immune response.
There is abundant epidemiological and experimental evidence that aspirin and other NSAIDs can inhibit tumor development in a number of organs, and such drugs have given positive results in human intervention studies. Considering the severe side effects of NSAIDs, inhibitors of mPGES-1 could be a better option for the treatment of patients with brain cancer.
Production of PGE2 in the brain is a critical step for neuron–glia–endothelial cell communications that underlie physiological and pathological processes in the brain. Inhibition of PGE2 production by NSAIDs is effective in ameliorating symptoms of inflammation. However, the gastrointestinal and cardiovascular side effects associated with COX-1 and COX-2 inhibition, respectively, have limited their use. Studies of mPGES-1 deficient mice provide compelling evidence for its role in a variety of inflammatory brain diseases and provide clues for developing new therapeutic treatment, because mPGES-1 may be a promising common target for several inflammatory brain diseases (Fig. 3). Indeed, inhibitors of mPGES-1 are being developed in light of that might provide the benefits of selective COX-2 inhibitors without the cardiovascular risk ascribed to inhibition of endothelial prostacyclin.11) Despite the fact that a number of mPGES-1 inhibitors have been generated over a long period of time, there are currently no selective mPGES-1 inhibitors available for clinical use. Recently, dual human/rodent mPGES-1 specific inhibitors have been reported, which revealed previously unknown differences between the effects of genetic disruption of mPGES-1 and pharmacological inhibition of mPGES-1 in in vivo models of inflammation.49) Therefore, the improvement of clinical symptoms of inflammatory brain diseases reported previously by using mPGES-1 deficient mice, as well as potentially severe side effects, should be reinvestigated using potent mPGES-1 specific inhibitors. Future clinical studies will address the important question of the efficacy and safety of mPGES-1 inhibition in human brain diseases.
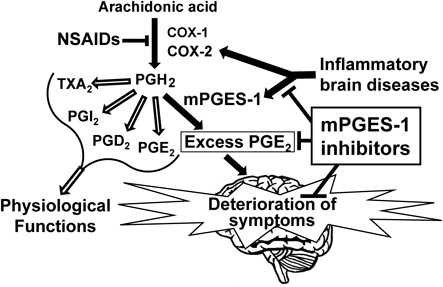
The excess PGE2 produced by induction of COX-2 and mPGES-1 in the lesion site of the brain contributes to deterioration of symptoms of inflammatory brain diseases. mPGES-1 inhibitors are supposed to specifically inhibit the pathological process and to not affect the physiological functions of constitutively existing prostanoids, while NSAIDs non-specifically suppress prostanoids that are important for physiological functions.
The author is grateful to Professor Norio Matsuki of Tokyo University and Professors Sachiko Oh-ishi, Yasuharu Sasaki and Mitsuo Tanabe of Kitasato University for their advice and encouragement. This study was supported in part by a JPSP KAKENHI (Grant Number 18790063), The Mochida Memorial Foundation for Medical and Pharmaceutical Research, The Uehara Memorial Foundation and Kitasato University Research Grant for Young Researchers.
The author declares no conflict of interest.