MATERIALS AND METHODS
MaterialsAuthentic standards of fentanyl, acetylfentanyl, and their metabolites were synthesized in our laboratory. cis-3-Methylfentanyl was synthesized in our laboratory by the method reported previously.9) β-Glucuronidase/aryl sulfatase (from Helix pomatia; β-glucuronidase, 32 units/mL; aryl sulfatase, 102 units/mL) was purchased from Merck (Darmstadt, Germany). h-iPS-HEP (Cellartis™ enhanced h-iPS-HEP, differentiated from ChiPSC18 and ChiPSC22), Cellartis HEP Coat, and Cellartis HEP supplement were purchased from TaKaRa Bio Inc. (Kusatsu, Japan). h-iPS-HEP (ReproHepato™), ReproHepato culture medium, and ReproHepato assay medium were purchased from ReproCELL Inc. (Yokohama, Japan). h-PRM-HEP and KLC-SuM medium were purchased from Kurabo Industries (Osaka, Japan). InVitroGRO CP medium and InVitroGRO HT medium were purchased from BioreclamationIVT (New York, NY, U.S.A.). Y-27632 was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Matrigel™ was purchased from Corning (Corning, NY, U.S.A.). Phosphate-buffered saline (PBS), Williams Medium E and Leibovitz’s L-15 medium were purchased from Thermo Fisher Scientific (Waltham, MA, U.S.A.). All other reagents used were of analytical grade.
Synthesis of Authentic Standards of Drugs and MetabolitesAll synthesized standards were confirmed by positive electrospray ionization (ESI) mass spectrometry and 1H-NMR. ESI mass spectra were obtained from a LCQ FLEET ion trap mass spectrometer (Thermo Fisher Scientific). 1H-NMR spectra were measured on a JNM-ECA600 NMR spectrometer (JEOL, Akishima, Japan). Tetramethylsilane was used as an internal standard.
N-Phenyl-N-[1-(2-phenylethyl)-4-piperidinyl]propanamide (Fentanyl)Fentanyl was synthesized according to the method of Siegfried.10) Briefly, 1-(2-phenylethyl)-4-piperidone and aniline were condensed in the presence of 3 Å molecular sieves. The product was reduced by sodium borohydride to give N-phenyl-1-(2-phenylethyl)-4-piperidinamine (despropionyl-fentanyl, 0.897 g, 65% yield from 1 g of 1-(2-phenylethyl)-4-piperidone), and this was then propionylated with propionyl chloride to give fentanyl. The free base of fentanyl was converted to the hydrochloride salt using hydrochloric acid (84 mg, 63% yield from 100 mg of despropionyl-fentanyl). Acetylfentanyl (N-phenyl-N-[1-(2-phenylethyl)-4-piperidinyl]acetamide) hydrochloride was synthesized by the same method (90 mg, 70% yield from 100 mg of despropionyl-fentanyl).
Fentanyl (free base)—1H-NMR (CDCl3) δ: 1.02 (3H, t, J=7.3 Hz), 1.96 (2H, q, J=7.3 Hz), 1.97–2.02 (2H, m), 2.18 (2H, qd, J=3.3 Hz, 13.0 Hz), 2.80 (2H, qd, J=2.2 Hz, 11.6 Hz), 3.08–3.13 (2H, m), 3.19–3.23 (2H, m), 3.60 (2H, br d, J=10.8 Hz), 4.79 (1H, tt, J=3.8 Hz, 12.3 Hz), 7.08–7.12 (2H, m), 7.20–7.32 (5H, m), 7.40–7.49 (3H, m).
Acetylfentanyl (free base)—1H-NMR (CDCl3) δ: 1.78 (3H, s), 1.98–2.02 (2H, m), 2.20 (2H, qd, J=3.3 Hz, 13.1 Hz), 2.80 (2H, qd, J=2.1 Hz, 11.7 Hz), 3.07–3.13 (2H, m), 3.19–3.23 (2H, m), 3.60 (2H, br d, J=10.8 Hz), 4.78 (1H, tt, J=3.5 Hz, 12.2 Hz), 7.08–7.12 (2H, m), 7.20–7.33 (5H, m), 7.40–7.49 (3H, m).
N-Phenyl-N-(4-piperidinyl)propanamide (Nor-fentanyl)1-Benzyl-4-piperidone and aniline were condensed in the presence of 3 Å molecular sieves, followed by reduction with sodium borohydride and propionylation with propionyl chloride. Debenzylation of the product by catalytic hydrogenation under acidic conditions (hydrochloric acid in methanol) using 5% palladium on carbon as a catalyst gave nor-fentanyl hydrochloride (352 mg, 70% yield from 356 mg of 1-benzyl-4-piperidone). Nor-acetylfentanyl (N-phenyl-N-(4-piperidinyl)acetamide) hydrochloride was synthesized by the same method (286 mg, 69% yield from 309 mg of 1-benzyl-4-piperidone).
Nor-fentanyl hydrochloride—1H-NMR (CD3OD) δ: 0.99 (3H, t, J=7.2 Hz), 1.59 (2H, qd, J=4.2 Hz, 13.0 Hz), 1.98 (2H, q, J=7.2 Hz), 2.08 (2H, br d, J=13.8 Hz), 3.11 (2H, td, J=2.6 Hz, 13.4 Hz), 3.36–3.42 (2H, m), 4.77 (1H, tt, J=3.6 Hz, 12.6 Hz), 7.23–7.27 (2H, m), 7.46–7.54 (3H, m).
Nor-acetylfentanyl hydrochloride—1H-NMR (CD3OD) δ: 1.60 (2H, qd, J=4.0 Hz, 13.0 Hz), 1.76 (3H, s), 2.09 (2H, br d, J=13.8 Hz), 3.10 (2H, td, J=3.0 Hz, 13.5 Hz), 3.36–3.43 (2H, m), 4.77 (1H, tt, J=3.6 Hz, 12.6 Hz), 7.25–7.30 (2H, m), 7.47–7.56 (3H, m).
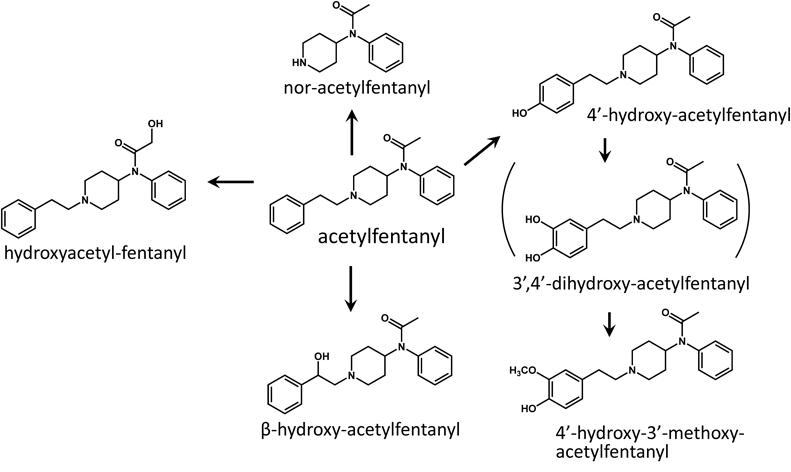
N-Phenyl-N-[1-[2-(4-hydroxyphenyl)ethyl]-4-piperidinyl]propanamide (4′-Hydroxy-fentanyl)4-Benzyloxyphenylacetic acid was reduced by lithium aluminum hydride to give 4-benzyloxyphenylethyl alcohol. 4-Benzyloxyphenylethyl alcohol was treated with methanesulfonyl chloride to convert the alcohol to the methansulfonic acid ester.11) Condensation of the ester with nor-fentanyl,12) followed by debenzylation gave 4′-hydroxy-fentanyl hydrochloride (34 mg, 59% yield from 40 mg of nor-fentanyl hydrochloride). 4′-Hydroxy-acetylfentanyl (N-phenyl-N-[1-[2-(4-hydroxyphenyl)ethyl]-4-piperidinyl]acetamide) hydrochloride (24 mg, 41% yield from 40 mg of nor-acetylfentanyl hydrochloride), 4′-hydroxy-3′-methoxy-fentanyl (N-phenyl-N-[1-[2-(4-hydroxy-3-methoxyphenyl)ethyl]-4-piperidinyl]propanamide) hydrochloride (26 mg, 42% yield from 40 mg of nor-fentanyl hydrochloride), and 4′-hydroxy-3′-methoxy-acetylfentanyl (N-phenyl-N-[1-[2-(4-hydroxy-3-methoxyphenyl)ethyl]-4-piperidinyl]acetamide) hydrochloride (28 mg, 44% yield from 40 mg of nor-acetylfentanyl hydrochloride) were synthesized by the same method.
4′-Hydroxy-fentanyl (free base)—1H-NMR (CDCl3) δ: 1.01 (3H, t, J=7.1 Hz), 1.96 (2H, q, J=7.1 Hz), 1.98–2.05 (2H, m), 2.11–2.21 (2H, m), 2.74–2.84 (2H, m), 3.01–3.16 (4H, m), 3.55–3.64 (2H, m), 4.74–4.84 (1H, m), 6.78 (2H, d, J=8.7 Hz), 7.03 (2H, d, J=8.7 Hz), 7.09 (2H, d, J=7.2 Hz), 7.40–7.50 (3H, m).
4′-Hydroxy-acetylfentanyl (free base)—1H-NMR (CDCl3) δ: 1.78 (3H, s), 1.97–2.06 (2H, m), 2.13–2.22 (2H, m), 2.74–2.84 (2H, m), 3.00–3.15 (4H, m), 3.55–3.63 (2H, m), 4.74–4.83 (1H, m), 6.79 (2H, d, J=7.8 Hz), 7.02 (2H, d, J=7.8 Hz), 7.10 (2H, d, J=6.6 Hz), 7.40–7.49 (3H, m).
4′-Hydroxy-3′-methoxy-fentanyl (free base)—1H-NMR (CDCl3) δ: 1.02 (3H, t, J=7.5 Hz), 1.96 (2H, q, J=7.5 Hz), 1.98–2.05 (2H, m), 2.18 (2H, qd, J=3.7 Hz, 13.0 Hz), 2.75–2.85 (2H, m), 3.03–3.17 (4H, m), 3.59 (2H, br d, J=12.0 Hz), 3.87 (3H, s), 4.79 (1H, tt, J=3.5 Hz, 12.2 Hz), 6.66 (1H, dd, J=1.5 Hz, 7.8 Hz), 6.77 (1H, d, J=1.5 Hz), 6.83 (1H, d, J=7.8 Hz), 7.09–7.13 (2H, m), 7.40–7.50 (3H, m).
4′-Hydroxy-3′-methoxy-acetylfentanyl (free base)—1H-NMR (CDCl3) δ: 1.78 (3H, s), 1.98–2.06 (2H, m), 2.19 (2H, qd, J=3.5 Hz, 13.1 Hz), 2.75–2.84 (2H, m), 3.03–3.17 (4H, m), 3.59 (2H, br d, J=10.8 Hz), 3.87 (3H, s), 4.78 (1H, tt, J=3.4 Hz, 12.2 Hz), 6.66 (1H, dd, J=1.8 Hz, 8.1 Hz), 6.77 (1H, d, J=1.8 Hz), 6.83 (1H, d, J=8.1 Hz), 7.09–7.12 (2H, m), 7.40–7.50 (3H, m).
N-Phenyl-N-[1-(2-phenylethyl)-4-piperidinyl]-3-hydroxypropanamide (ω-Hydroxy-fentanyl)To a solution of 96 mg of 3-benzyloxypropionic acid in 1 mL of toluene, 2 drops of N,N-dimethylformamide and 129 mg of thionyl chloride were added and the mixture was heated at 90°C for 1.5 h. The reaction mixture was added dropwise to a solution of 30 mg of despropionyl-fentanyl in 1 mL of pyridine. After the addition, water was added to the reaction mixture, the mixture was acidified with 3 M hydrochloric acid, then extracted with chloroform. The extract was purified by flash chromatography (column, silica gel; solvent, n-hexane/ethyl acetate). Debenzylation of the product by catalytic hydrogenation under acidic conditions using 5% palladium on carbon as a catalyst gave 25 mg of ω-hydroxy-fentanyl hydrochloride (60% yield from despropionyl-fentanyl).
ω-Hydroxy-fentanyl (free base)—1H-NMR (CDCl3) δ: 1.98–2.03 (2H, m), 2.18 (2H, t, J=5.4 Hz), 2.23 (2H, qd, J=3.5 Hz, 13.3 Hz), 2.75–2.84 (2H, m), 3.08–3.14 (2H, m), 3.19–3.24 (2H, m), 3.61 (2H, br d, J=11.4 Hz), 3.74 (2H, t, J=5.4 Hz), 4.78 (1H, tt, J=3.4 Hz, 12.2 Hz), 7.09–7.14 (2H, m), 7.20–7.33 (5H, m), 7.42–7.50 (3H, m).
N-Phenyl-N-[1-(2-phenylethyl)-4-piperidinyl]-2-hydroxypropanamide ((ω-1)-Hydroxy-fentanyl)To a solution of 50 mg of despropionyl-fentanyl in 2 mL of dichloromethane, 40 µL of triethylamine, and 40.3 mg of 2-acetoxypropionyl chloride were added and stirred for 2 h at room temperature. To the reaction mixture, 20 mL of 0.1 M hydrochloric acid was added, and then extracted with chloroform. The solvent was evaporated to dryness under vacuum and the residue was treated with a small amount (ca. 15 drops) of 1 M sodium hydroxide in methanol for 5 min to remove the acetyl group. The solution was neutralized with 1 M acetic acid in methanol, then water was added and the solution was basified with 28% aqueous ammonia, then extracted with chloroform. The residue was recrystallized from methanol to give 41 mg of (ω-1)-hydroxy-fentanyl (64% yield from despropionyl-fentanyl). Hydroxyacetyl-fentanyl (N-phenyl-N-[1-(2-phenylethyl)-4-piperidinyl]-2-hydroxyacetamide) (free base) was synthesized by the same method (16 mg, 27% yield from 50 mg of despropionyl-fentanyl).
(ω-1)-Hydroxy-fentanyl (free base)—1H-NMR (CDCl3) δ: 1.09 (3H, d, J=6.5 Hz), 1.87–1.93 (1H, m), 2.09–2.14 (1H, m), 2.18 (1H, qd, J=3.9 Hz, 13.3 Hz), 2.36 (1H, qd, J=4.2 Hz, 13.2 Hz), 2.75–2.85 (2H, m), 3.08–3.14 (2H, m), 3.19–3.25 (2H, m), 3.57–3.66 (2H, m), 4.75 (1H, tt, J=3.7 Hz, 12.2 Hz), 7.11–7.33 (7H, m), 7.45–7.53 (3H, m).
Hydroxyacetyl-fentanyl (free base)—1H-NMR (CDCl3) δ: 2.04 (2H, br d, J=13.8 Hz), 2.28 (2H, qd, J=3.6 Hz, 12.9 Hz), 2.77–2.86 (2H, m), 3.09–3.14 (2H, m), 3.20–3.24 (2H, m), 3.60–3.64 (2H, m), 3.70 (2H, s), 4.76 (1H, tt, J=3.4 Hz, 12.3 Hz), 7.10–7.14 (2H, m), 7.20–7.33 (5H, m), 7.47–7.52 (3H, m).
N-Phenyl-N-[1-(2-hydroxy-2-phenylethyl)-4-piperidinyl]propanamide (β-Hydroxy-fentanyl)A mixture of 40 mg of nor-fentanyl hydrochloride, 28 mg of phenacyl chloride, and 15 mg of sodium bicarbonate in 1.2 mL of water–2-butanone (1 : 5) was heated at 90°C for 6 h. Water was added to the reaction mixture, and the mixture was basified with 28% aqueous ammonia, then extracted with ethyl acetate. The extract was purified by flash chromatography (column, silica gel; solvent, chloroform/ethyl acetate). The purified product, β-hydroxy-fentanyl was converted to the hydrochloride salt (18 mg, 31% yield from 40 mg of nor-fentanyl hydrochloride). β-Hydroxy-acetylfentanyl (N-phenyl-N-[1-(2-hydroxy-2-phenylethyl)-4-piperidinyl]acetamide) hydrochloride was synthesized by the same method (34 mg, 58% yield from 40 mg of nor-acetylfentanyl hydrochloride).
β-Hydroxy-fentanyl (free base)—1H-NMR (CDCl3) δ: 1.02 (3H, t, J=7.3 Hz), 1.97 (2H, q, J=7.3 Hz), 1.98–2.02 (1H, m), 2.04–2.10 (1H, m), 2.19 (1H, qd, J=3.7 Hz, 13.0 Hz), 2.27 (1H, qd, J=3.9 Hz, 13.1 Hz), 2.88–2.98 (2H, m), 2.98–3.04 (1H, m), 3.16–3.23 (1H, m), 3.79–3.86 (2H, br), 4.83 (1H, tt, J=3.8 Hz, 12.4 Hz), 5.34 (1H, dd, J=2.4 Hz, 10.2 Hz), 7.02–7.13 (2H, br), 7.28–7.33 (1H, m), 7.33–7.38 (4H, m), 7.41–7.50 (3H, m).
β-Hydroxy-acetylfentanyl (free base)—1H-NMR (CDCl3) δ: 1.79 (3H, s), 1.97–2.03 (1H, m), 2.05–2.10 (1H, m), 2.21 (1H, qd, J=3.9 Hz, 13.1 Hz), 2.29 (1H, qd, J=3.7 Hz, 13.3 Hz), 2.88–2.98 (2H, m), 2.98–3.04 (1H, m), 3.16–3.22 (1H, m), 3.79–3.86 (2H, br), 4.82 (1H, tt, J=3.5 Hz, 12.3 Hz), 5.35 (1H, dd, J=1.8 Hz, 10.2 Hz), 7.10–7.14 (2H, br), 7.28–7.33 (1H, m), 7.33–7.38 (4H, m), 7.41–7.50 (3H, m).
Incubation of Drugs with h-iPS-HEP (Cellartis™)Cellartis enhanced h-iPS-HEP (≥1.23×107 viable cells/vial, from ChiPSC18 or ChiPSC22) were thawed in 40 mL of warm InVitroGRO HT medium that contained 5 µM of Y-27632. After centrifugation at 100×g for 2 min, the supernatant was removed, then the cells were resuspended in 15 mL of warm InVitroGRO CP medium that contained 5 µM of Y-27632. A 1 mL portion of the cell suspension was dispensed into each well of a 24-well microplate coated with Cellartis HEP Coat and incubated at 37°C and 5% CO2. After 1, 3, and 5 d from the start of incubation, the medium was changed to 0.5 mL of warm fresh Williams Medium E that contained 2% Cellartis HEP supplement solution and 0.5% dimethyl sulfoxide. After changing the medium at day 5, the drug (fentanyl hydrochloride or acetylfentanyl hydrochloride dissolved in PBS) was added to each well of the plate at a final concentration of 10 µM, and incubated for 24 or 48 h. The medium was collected and stored at −30°C until analysis.
Incubation of Drugs with h-iPS-HEP (ReproHepato™)ReproHepato h-iPS-HEP (8.2×106 cells/vial) was thawed in 49 mL of warm Leibovitz’s L-15 medium. After centrifugation at 350×g for 5 min, the supernatant was removed, then the cells were resuspended in 11 mL of warm ReproHepato culture medium. A 640 µL portion of the cell suspension was dispensed into each well of a 24-well microplate coated with Matrigel, and incubated at 37°C and 5% CO2. After 1, 3, and 5 d from the start of incubation, the medium was changed to 0.5 mL of warm fresh ReproHepato culture medium. After 6 d from the start of incubation, the medium was changed to 0.5 mL of warm fresh ReproHepato assay medium, then the drug (fentanyl hydrochloride or acetylfentanyl hydrochloride dissolved in PBS) was added to each well of the plate at a final concentration of 10 µM, and incubated for 24 or 48 h. The medium was collected and stored at −30°C until analysis.
Incubation of Drugs with h-PRM-HEPh-PRM-HEP (2×106 viable cells/vial) was thawed in 20 mL of warm Leibovitz’s L-15 medium. After centrifugation at 170×g for 2 min, the supernatant was removed, then the cells were resuspended in 2 mL of warm KLC-SuM medium and the cell viability was measured by the trypan blue dye exclusion method. The cell suspension was diluted to 5×105 viable cells/mL with KLC-SuM medium and 0.5 mL of the cell suspension was dispensed into each well of a 24-well microplate. The drug (fentanyl hydrochloride or acetylfentanyl hydrochloride dissolved in PBS) was added to each well of the plate at a final concentration of 10 µM, and incubated for 3 h with shaking (90 rpm) at 37°C and 5% CO2. The medium was collected and stored at −30°C until analysis.
Identification of the MetabolitesTo a 25 µL sample of the medium, 15 µL of 0.25 M acetate buffer (pH 5.0) containing β-glucuronidase/aryl sulfatase (β-glucuronidase, 0.01 unit) was added and then incubated at 60°C for 1.5 h. To the reaction mixture, 250 µL of acetonitrile was added and the mixture was vortexed for 5 s. After centrifugation at 10000×g for 5 min, the supernatant was evaporated to dryness under a nitrogen stream. The residue was reconstituted with 50 µL of the initial mobile phase and centrifuged at 10000×g for 5 min, then the supernatant (10 µL) was analyzed by LC-ion trap MS under scan and product ion analysis modes. The conditions of analysis were as follows: apparatus, Accela LC system connected to LCQ FLEET ion trap mass spectrometer (Thermo Fisher Scientific); column, CORTECS C18 (2.1×50 mm, 2.7 µm, Waters, Milford, MA, U.S.A.) maintained at 40°C; mobile phase composition, 0.1% formic acid (A) and methanol (B); linear gradient mode, 20% B for 1 min, 20% to 80% B over 8 min, 80% B for 2 min, and 80% to 20% B over 0.1 min; flow rate, 0.2 mL/min; MS interface, positive ESI; analysis mode, scan (m/z 100–500) and product ion analysis (normalized collision energy, 35%; precursor ions, protonated molecules of drugs and putative metabolites).
Quantitation of the MetabolitesTo a 25 µL sample of the medium, 15 µL of 0.25 M acetate buffer (pH 5.0) containing β-glucuronidase/aryl sulfatase (β-glucuronidase, 0.01 unit) was added and then incubated at 60°C for 1.5 h. To the reaction mixture, 10 µL of internal standard solution (cis-3-methylfentanyl hydrochloride, 50 ng/10 µL water) was added, then 250 µL of acetonitrile was added and the mixture was vortexed for 5 s. After centrifugation at 10000×g for 5 min, 50 µL of the supernatant was mixed with 200 µL of 0.1% formic acid. This was centrifuged at 10000×g for 5 min, then the supernatant (10 µL) was analyzed by LC-triple quadrupole MS. The conditions of analysis were as follows: apparatus, NANOSPACE SI-2 LC system (Shiseido, Tokyo, Japan) connected to TSQ Quantum triple quadrupole mass spectrometer (Thermo Fisher Scientific); column, mobile phase composition, flow rate, and MS interface were the same as for identification of the metabolites; analysis mode, selected reaction monitoring (SRM). SRM parameters are listed in Table 1.
Table 1. Monitoring Ion, Collision Energy and Calibration Curve for Analysis of Compounds by Liquid Chromatography-Tandem Mass Spectrometry
| Compound | Monitoring ion (m/z) | Collision energy (eV) | Calibration curve (µM) |
|---|
| Precursor ([M+H]+) | Product |
|---|
| Fentanyl | 337.1 | 188.2 | 21 | 0.021–11 |
| Nor-fentanyl | 233.1 | 84.2 | 17 | 0.030–15 |
| ω-Hydroxy-fentanyl | 353.1 | 188.2 | 21 | 0.021–5.1 |
| (ω-1)-Hydroxy-fentanyl | 353.1 | 188.2 | 20 | 0.023–5.7 |
| 4′-Hydroxy-fentanyl | 353.1 | 121.1 | 32 | 0.021–5.1 |
| β-Hydroxy-fentanyl | 353.1 | 204.2 | 19 | 0.021–5.1 |
| 4′-Hydroxy-3′-methoxy-fentanyl | 383.1 | 151.2 | 30 | 0.019–4.8 |
| Acetylfentanyl | 323.1 | 188.2 | 20 | 0.022–11 |
| Nor-acetylfentanyl | 219.1 | 84.2 | 18 | 0.031–16 |
| Hydroxyacetyl-fentanyl | 339.1 | 188.2 | 20 | 0.024–5.9 |
| 4′-Hydroxy-acetylfentanyl | 339.1 | 121.1 | 31 | 0.021–5.3 |
| β-Hydroxy-acetylfentanyl | 339.1 | 321.3 | 16 | 0.021–5.3 |
| 4′-Hydroxy-3′-methoxy-acetylfentanyl | 369.1 | 151.2 | 28 | 0.020–4.9 |
| cis-3-Methylfentanyl (Internal standard) | 351.1 | 202.2 | 24 | — |
Authentic standards of fentanyl, acetylfentanyl, and their metabolites were added to the InVitroGRO CP medium and processed as described above to obtain the calibration curves. Excellent linearity was obtained over the concentration range 0.008–4 µg/mL (e.g. 0.021–11 µM for fentanyl hydrochloride, Table 1) for fentanyl, acetylfentanyl, nor-fentanyl, and nor-acetylfentanyl and 0.008–2 µg/mL for the other compounds, with a correlation coefficient of 0.99.