Abstract
Avian influenza A (H7N9) virus has caused several epidemics and infection in both human and poultry. With mutation, the H7N9 virus gained its fifth endemic in China. Early diagnosis is crucial for the control of viral spread in poultry and prognosis of infected patients. In this study, we developed and evaluated a lateral flow dipstick recombinase polymerase amplification (LFD-RPA) assay for rapid detection of both hemagglutinin and neuraminidase gene of H7N9. Our H7-LFD-RPA and N9-LFD-RPA assay were able to detect 32 fg H7N9 nucleic acid which is more convenient and rapid than previous methods. Through detecting 50 influenza positive samples, cross-reaction was not found with other subtypes of influenza virus. The 100% analytical specificity and sufficient analytical sensitivity results agreed the real time RT-PCR assay. The results data demonstrated that our method performed well and could be applied to the detection of H7N9 virus. This LFD-RPA assay provides a candidate method for rapid point-of-care diagnosis of H7N9.
Avian influenza A (H7N9) virus has caused five epidemics in China, the first human case was identified in March 2013.1,2) Until December 30th 2017, a total of 1564 cases from 25 provinces in China were diagnosed with H7N9 infection and the mortality is up reaching 40%. The number of cases in the fifth endemic has an unparalleled increase,3) accounting for nearly half of the total cases. The first four waves of the epidemic demonstrated that the virus had low pathogenicity in birds. However, insertion of four amino acid in the hemagglutinin (HA) protein was found in the fifth wave H7N9 strains isolated from some patients, live poultry markets and environmental samples, which means the H7N9 might be mutated to be highly pathogenic,4) and thus resulted in serious economic losses as well as an international concern.2)
Early identification of H7N9 positive patients and poultry is crucial for disease control and reduction of economic loss. The existing diagnostic methods for H7N9, such as viral isolation and enzyme-linked immunosorbent assay (ELISA), have comparatively low sensitivity.5,6) Real time RT-PCR, which is accurate and rapid (90 min of a singly run-time), has become a routine detection for years.7) Nevertheless, RT-PCR requires sophisticated equipment restricted to the laboratory for point-of-care diagnosis of patients with flu symptom in hospital and samples in poultry market, etc. Recently, recombinase polymerase amplification (RPA), one of the most sensitive, convenient and rapid isothermal amplification methods, was developed for the detection variety of pathogens.8–10) Moreover, lateral flow dipstick (LFD), a simple device, has been used in non-laboratory environment. An established visual detection system by combining RPA and LFD has demonstrated satisfying applications in some pathogenic detection,6,11,12) however there is no LFD-RPA assay developed for influenza virus detection. Thus in this research, we developed and evaluated a rapid detection method based on LFD-RPA assay for both HA and neuraminidase (NA) gene of avian influenza A (H7N9).
MATERIALS AND METHODS
Sample CollectionPositive and control viruses used in this study were provided by Shenzhen Center for Disease Control and Prevention (Shenzhen CDC). Clinical specimens and environmental specimens, including swabs and serum within three days of onset after disease, were collected in Shenzhen, China. Positive samples were confirmed by One Step PrimerScript™ RT-PCR kit (TaKaRa, Japan).
Virus RNA ExtractionThe RNA isolated from a H7N9 infected patient, which was confirmed by laboratory assay and provided by Shenzhen CDC, was used as template in the RPA reaction and sensitivity analysis. RNA extraction via magnetic bead method (Life Technologies, Darmstadt, Germany) according to the manufacturer’s instructions. In summary, 200 µL of sample was incubated with 50 µL of proteinase K and 300 µL of lysis buffer for 5 min at room temperature. Thereafter, 150 µL of isopropanol and 50 µL of Dynabeads were added and incubated for 10 min followed by two washing steps. Nucleic acid bound to the Dynabeads was dried for 5 min, followed by elution. The total time needed for the extraction was 30 min. The extracted influenza virus RNA was first reversely transcribed into cDNA with PrimerScript™ 1st Strand cDNA Synthesis Kit (TaKaRa). In summary, 8 µL of template RNA was incubated with 1 µL of Oligo dT Primer and 1 µL of deoxyribonucleoside triphosphate (dNTP). Mixture for 5 min at 65°C, cool immediately on ice. Thereafter, 4 µL of 5×PrimeScript II Buffer and 0.5 µL of Ribonuclease (RNase) Inhibitor and 1 µL of PrimeScript II RTase and 4.5 µL of RNase free dH2O were added and incubated for 50 min at 42°C. Terminate the reactions at 70°C for 15 min, followed by cooling on ice. Concentration of the cDNA was 1400 ng/µL measured by q-RT-PCR and quantified to 1000 ng/µL. Transcribed cDNA was stored at −20°C used for further analysis.
H7 and N9 RPA Primers and Probes DesignBased on the sequences of highly conserved and specific domains of H7N9 avian influenza viruses, which were randomly chosen in GenBank, three pairs of primers and probes for H7 and N9 gene were designed by Primer 5.0. Primers and probes for H7 and N9 were separately aligned with the HA and NA gene consensus sequences of H7N9 viruses. The opposing amplification primers were conjoined with a biotin-label on the 5′ end and the probes contained a 5′ FAM (carboxyfluorescein)-label, an internal tetrahydrofuran (THF) and a polymerase extension blocking group, C3-spacer at the 3′ end. All oligonucleotides were produced by Sangon Biotech (Shenzhen, China).
RPA Assay and ConditionsThe RPA assay was firstly performed with the TwistAmp nfo kit (TwistDX, Cambridge, U.K.) in a volume of 50 µL, containing 29.5 µL of rehydration buffer, 3.2 µL of template,10 µL of dH2O, 0.6 µL of probe nfo-H7-P or nfo-N9-P (10 µM), 2.1 µL of forward primer (10 µM), 2.1 µL of biotin-labeled reverse primer (10 µM) and 2.5 µL of magnesium acetate solution and the mixture was added to RPA strips containing a dried enzyme pellet.
All the three sets of primers and probes were used to perform RPA for efficiency and specificity evaluation, the best set was kept for later study. Proper amplification time and temperature were also explored according to the result of real-time RPA assay performed with TwistAmp exo kit (TwistDX, Cambridge, U.K.) on 7500 Real Time PCR System (Applied Biosystems, MA, U.S.A.) according to the previous studies.10,13)
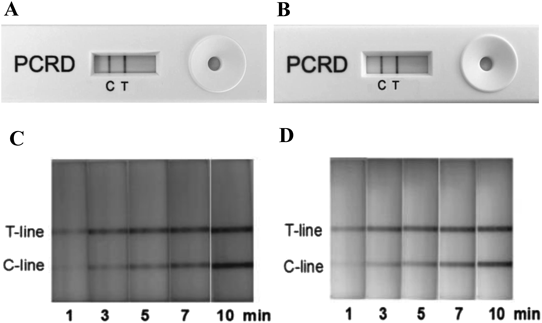
LFD RPA AssayFive microliters of the amplification product and 70 µL of the PCRD Extraction Buffer (Abingdon Health, U.K.) were mixed thoroughly and added into the sample well of a PCRD cassette (Abingdon Health) for signal reading. Independent working areas and pipettors were utilized to avoid contamination. Upon sample amplification, the carbon conjugated-biotin antibodies are rehydrated and react with the biotin-labelled amplicon. The mixture travels along the membrane by capillary effect. Negative result is indicated by the presence of a single black line on the test strip (C-line) and positive result is recognized by that of both C and T-line position, respectively.
Evaluation of LFD-RPA and Statistical AnalysisA total of 50 samples (21 positive and 29 negative) were collected from both of swabs of H7N9 infected patients and the external environment. The samples were used to evaluate the efficiency of LFD-RPA method. The screening parameters, including sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) were used to evaluate the diagnostic validity. Kappa test was applied to verify the strength of agreement between two detection methods, RT-PCR and LFD-RPA.
RESULTS
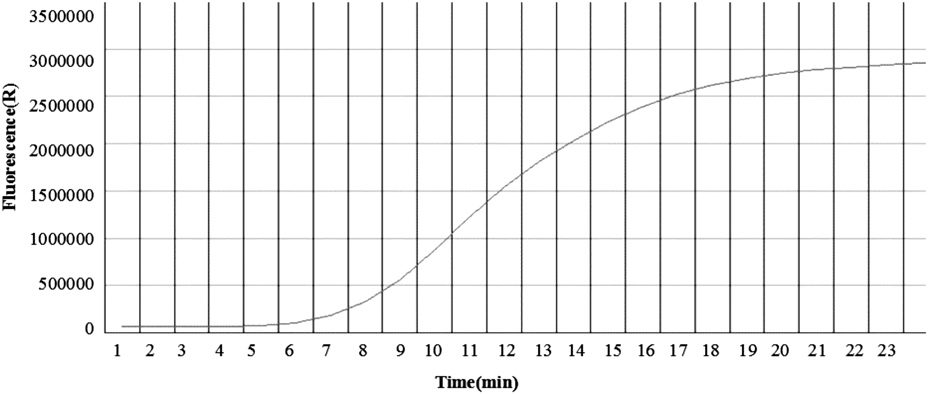
Optimal ConditionsThe sequences of primers and probes used for RPA assay are displayed in Table 1 and the optimal reaction conditions were 20 min at 39°C for both of H7 and N9 RPA assay(Fig. 1).
Table 1. LFD-RPA Primers and Probes for H7 and N9 Used in This Study
| Primer/probe | Sequence | Length (bp) | Product size (bp) |
|---|
| nfo-H7-F | TTCTATGCAGAAATGAAATGGCTCCTGTCAA | 31 | 96 |
| nfo-H7-R | [Biotin]AGCTGGGCTTTTTCTTGTATTTTTATATGACTTAG | 43 |
| nfo-H7-P | [FAM]AAATGAAATGGCTCCTGTCAAACACAGATA[THF]TGCTGCATTCCCGCA[C3-spacer] | 66 |
| nfo-N9-F | ATAGACCCAGTAGCAATGACACACACTAGTCA | 32 | 105 |
| nfo-N9-R | [Biotin]TTATTATTACCTGGATAAGGGTCATTACACT | 39 |
| nfo-N9-P | [FAM]CACTAGTCAATATATATGCAGTCCTGTTCTA[THF]AGACAGTCCCCGACCGA[C3-spacer] | 69 |
The positive results of lateral-flow cassette for H7 and N9 can be observed within 1 min (Figs. 2A–D). To determine the sensitivity of the assay, cDNA template (1000 ng/µL H7N9 cDNA) was serially diluted by 1010 (3200 ng to 0.32 fg). The LFD could detect up to 10−8 dilution (32 fg, 4×103 copies) of both H7 and N9 (Figs. 3A, B). The resulting RPA amplicons were electrophoresed in 3% agarose gel and visualized under UV. A visible amplicon band was obtained up to 10−6 dilution and amplicons of the desired size were generated: 96 bp with H7 and 105 bp with N9 (Figs. 3C, D).
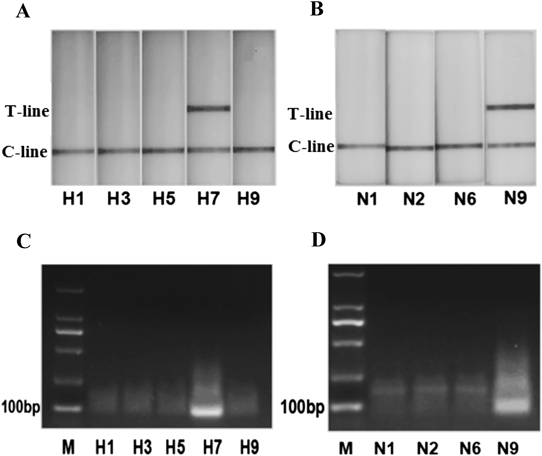
Analytical SpecificityThe LFD-RPA assay was tested for specificity using RNA extracted from other subtypes of influenza virus A which had been detected by RT-PCR assay. Only the H7 subtype detected by H7-LFD-RPA and the N9 subtype detected by N9-LFD-RPA were positive (Table 2). No cross-reaction was observed with other influenza virus of H1N1, H3N2, H5N6 and H9N2 (Figs. 4A, B), which were also confirmed by the agarose gel electrophoresis (Fig. 4C, D). No amplification was detected from H1, H3, H5 and H9 with H7-RPA or N1, N2 and N6 with N9-RPA.
Table 2. Detection of Multiple Subtypes by the LFD-RPA and RT-PCR Assays
| Subtype | H7 | N9 |
|---|
| PCR | RPA | PCR | RPA |
|---|
| H7N9 | + | + | + | + |
| H1N1 | − | − | − | − |
| H3N2 | − | − | − | − |
| H5N6 | − | − | − | − |
| H9N2 | − | − | − | − |
Total of 21 positive samples and 29 negative samples were determined by both H7-LFD-RPA and N9-LFD-RPA. This result showed the PPV and NPV of H7/N9-LFD-RPA reached 100%.
DISCUSSION
In order to efficiently detect the currently epidemic zoonosis disease H7N9, we successfully developed a visual method by combining an isothermal amplification technique with LFD-RPA. Through a 50-sample study, both H7-LFD-RPA and N9-LFD-RPA assay gained satisfying performances of 100% analytical sensitivity and analytical specificity. No cross-reaction with other subtypes of influenza virus was observed. Moreover, the results of multiple tests for the same sample indicated the method was biologically and technically repeatable.
Design of RPA primers and probes is the key and bottle-neck of RPA assay method. Previous study showed that RPA could amplify as long as 1.5 kb DNA segment with the best sensitivity and specificity at 100–200 bp.14) In this study, we chose the optimal sets of primers and probes for H7 and N9 respectively referred as Yehia’s method for H5N1 HA gene.14) The forward/reverse primers and nfo-probes were designed based on a total of 96 HA and 105 NA gene sequences respectively with 92.8 and 85.1% identity to H7 and N9 of whole H7N9 database from China. Moreover, we found that primers/probes of proper lengths were necessary for high specificity. As the length of primers/probes were longer than the PCR gene and the target gene was shorter than the PCR gene, the amplification of the target gene would be obtained with higher accuracy. This was similar to previous studies.11,15,16)
A unique real-time RPA was done to explore the role of incubation temperature and time because the brightness of the test bands appeared to change with these conditions.11,17) Runtime for our RPA is much shorter (20 min in LFD-RPA compared to 90 min in RT-PCR) owing to its shorter target gene to amplify. Like most RPA assays,13,18) body temperature or a simple water bath can be applied to fulfill the detection temperature of 39°C, which is rather suitable for point-of-care diagnosis and potential study of genetic expression in living cells. The LFD method developed here greatly simplifies the detection process and visualize the results without the input of expensive equipment.
The LFD-RPA method established in this study can be used for H7N9 detection of both environmental and clinical samples with analytical sensitivity as 32 fg for either H7 or N9. Moreover, satisfying specificity of our LFD-RPA method is observed in distinguishing H7N9 from other subtypes, such as H1N1, H3N2, H5N6 and H9N2 which causes similar clinical respiratory symptoms as the H7N9 infected patients or poultry.
In this study, for the first time, we developed a simple visual method to detect H7N9 by combining LFD with RPA assay. All the results suggested that our method was robust against H7N9. Our LFD-RPA method enabled rapid monitoring and detection of H7N9 in endemic areas. However, in the preliminary stage, this method could not distinguish the subtypes except H7 and N9 yet. Further researches are necessary to optimize the labels of the primer and LFD to achieve a concurrent detection for H7 and N9 in one dipstick and identify other specific subtypes. Moreover, as H10 is closely related to H7, the H10-positive samples should be considered for further optimization of LFD-RPA method in the future.
Acknowledgments
This work was supported by the Municipal Healthcare Joint-Innovation Major Project of Guangzhou (201604020011), the Guangzhou Planning Fund of Science and Technology (201704020056), the Basic Research Project of Shenzhen Science and Technology Innovation Commission (JCYJ20170306160244540), and the Project of Shenzhen Health and Family Planning Commission (SZFZ2017020).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Wang J, Wang J, Liu L, Yuan W. Development of a real-time recombinase polymerase amplification assay for rapid and sensitive detection of porcine circovirus 2. Arch. Virol., 162, 2293–2296 (2017).
- 2) Su S, Gu M, Liu D, Cui J, Gao GF, Zhou J, Liu X. Epidemiology, evolution, and pathogenesis of H7N9 influenza viruses in five Epidemic Waves since 2013 in China. Trends Microbiol., 25, 713–728 (2017).
- 3) Zhou L, Ren R, Yang L, Bao C, Wu J, Wang D, Li C, Xiang N, Wang Y, Li D, Sui H, Shu Y, Feng Z, Li Q, Ni D. Sudden increase in human infection with avian influenza A (H7N9) virus in China, September–December 2016. Western Pac. Surveill. Response J., 8, 6–14 (2017).
- 4) Iuliano AD, Jang Y, Jones J, Davis CT, Wentworth DE, Uyeki TM, Roguski K, Thompson MG, Gubareva L, Fry AM, Burns E, Trock S, Zhou S, Katz JM, Jernigan DB. Increase in human infections with Avian Influenza A (H7N9) virus during the fifth epidemic—China, October 2016–February 2017. MMWR Morb. Mortal. Wkly. Rep., 66, 254–255 (2017).
- 5) Zhang Y, Mao H, Yan J, Zhang L, Sun Y, Wang X, Chen Y, Lu Y, Chen E, Lv H, Gong L, Li Z, Gao J, Xu C, Feng Y, Ge Q, Xu B, Xu F, Yang Z, Zhao G, Han J, Guus K, Li H, Shu Y, Chen Z, Xia S. Isolation and characterization of H7N9 avian influenza A virus from humans with respiratory diseases in Zhejiang, China. Virus Res., 189, 158–164 (2014).
- 6) Yu Y, Zhang X, Zhao B, Sun Y, Zhang X, Bai T, Lu J, Li Z, Liu L, Wang D, Shu Y, Zhou J, Qin K. A sandwich ELISA for the detection of neuraminidase of avian influenza A (H7N9) virus. J. Virol. Methods, 247, 58–60 (2017).
- 7) Chen Y, Wang D, Zheng S, Shu Y, Chen W, Cui D, Li J, Yu H, Wang Y, Li L, Shang H. Rapid diagnostic tests for identifying avian influenza A (H7N9) virus in clinical samples. Emerg. Infect. Dis., 21, 87–90 (2015).
- 8) Abd El Wahed A, El-Deeb A, El-Tholoth M, Abd El Kader H, Ahmed A, Hassan S, Hoffmann B, Haas B, Shalaby MA, Hufert FT, Weidmann M. A portable reverse transcription recombinase polymerase amplification assay for rapid detection of foot-and-mouth disease virus. PLOS ONE, 8, e71642 (2013).
- 9) Sun K, Xing W, Yu X, Fu W, Wang Y, Zou M, Luo Z, Xu D. Recombinase polymerase amplification combined with a lateral flow dipstick for rapid and visual detection of Schistosoma japonicum. Parasit. Vectors, 9, 476 (2016).
- 10) Teoh BT, Sam SS, Tan KK, Danlami MB, Shu MH, Johari J, Hooi PS, Brooks D, Piepenburg O, Nentwich O, Wilder-Smith A, Franco L, Tenorio A, AbuBakar S. Early detection of dengue virus by use of reverse transcription-recombinase polymerase amplification. J. Clin. Microbiol., 53, 830–837 (2015).
- 11) Crannell ZA, Cabada MM, Castellanos-Gonzalez A, Irani A, White AC, Richards-Kortum R. Recombinase polymerase amplification-based assay to diagnose Giardia in stool samples. Am. J. Trop. Med. Hyg., 92, 583–587 (2015).
- 12) Tu PA, Shiu JS, Lee SH, Pang VF, Wang DC, Wang PH. Development of a recombinase polymerase amplification lateral flow dipstick (RPA-LFD) for the field diagnosis of caprine arthritis-encephalitis virus (CAEV) infection. J. Virol. Methods, 243, 98–104 (2017).
- 13) Yang Y, Qin X, Zhang W, Li Z, Zhang S, Li Y, Zhang Z. Development of an isothermal recombinase polymerase amplification assay for rapid detection of pseudorabies virus. Mol. Cell. Probes, 33, 32–35 (2017).
- 14) Yehia N, Arafa AS, Abd El Wahed A, El-Sanousi AA, Weidmann M, Shalaby MA. Development of reverse transcription recombinase polymerase amplification assay for avian influenza H5N1 HA gene detection. J. Virol. Methods, 223, 45–49 (2015).
- 15) Abd El Wahed A, Patel P, Faye O, Thaloengsok S, Heidenreich D, Matangkasombut P, Manopwisedjaroen K, Sakuntabhai A, Sall AA, Hufert FT, Weidmann M. Recombinase polymerase amplification assay for rapid diagnostics of dengue infection. PLOS ONE, 10, e0129682 (2015).
- 16) Euler M, Wang Y, Heidenreich D, Patel P, Strohmeier O, Hakenberg S, Niedrig M, Hufert FT, Weidmann M. Development of a panel of recombinase polymerase amplification assays for detection of biothreat agents. J. Clin. Microbiol., 51, 1110–1117 (2013).
- 17) Lillis L, Lehman D, Singhal MC, Cantera J, Singleton J, Labarre P, Toyama A, Piepenburg O, Parker M, Wood R, Overbaugh J, Boyle DS. Non-instrumented incubation of a recombinase polymerase amplification assay for the rapid and sensitive detection of proviral HIV-1 DNA. PLOS ONE, 9, e108189 (2014).
- 18) Wang X, Jiang H, Wu P, Uyeki TM, Feng L, Lai S, Wang L, Huo X, Xu K, Chen E, Wang X, He J, Kang M, Zhang R, Zhang J, Wu J, Hu S, Zhang H, Liu X, Fu W, Ou J, Wu S, Qin Y, Zhang Z, Shi Y, Zhang J, Artois J, Fang VJ, Zhu H, Guan Y, Gilbert M, Horby PW, Leung GM, Gao GF, Cowling BJ, Yu H. Epidemiology of avian influenza A H7N9 virus in human beings across five epidemics in mainland China, 2013–17: An epidemiological study of laboratory-confirmed case series. Lancet Infect. Dis., 17, 822–832 (2017).