Abstract
We show that a lectin like protein from the mushroom Agaricus bisporus (LSMT) is capable to permeate the epithelial monolayer barrier of the intestine ex vivo. The protein is not toxic or immunogenic upon prolonged administration and elevated dose in mice. Thus, it could be a candidate as a drug carrier for oral administration. However, its permeability should be tested after the protein has been modified, mimicking the condition in which it is used as a drug carrier. The protein was conjugated to captopril, the selected model of a Biopharmaceutical Classification System (BCS) class III drug, with high solubility but poor permeability. The drug was conjugated to LSMT that had been modified with 4-succinimidyloxycarbonyl-alpha-methyl-2-pyridyldithiotoluene (SMPT) as a linker. The success of LSMT modification was confirmed with TLC and MS; the latter also indicated the amount of captopril molecule linked. The modified LSMT could permeate through the intestinal monolayer barrier, and thus could be absorbed in the intestine after modification. The modified protein appears to remain stable after incubation in simulated gastrointestinal fluids. This pioneering work provides an essential basis for further development of the protein as a drug carrier for oral administration.
The light subunit of mushroom tyrosinase (LSMT) is an endogenous protein of Agaricus bisporus that was serendipitously discovered upon the structure elucidation of the tyrosinase (PPO3) complex.1) The protein is encoded by the orf239342 gene and its expression is not correlated to that of PPO3 nor the other tyrosinases in the mushroom despite its co-localization with the tyrosinase encoding genes in the chromosome.2) LSMT is not identified as a mushroom lectin, and its structure resembles that of proteins of the lectin family,1) in particular to the non-toxic non-hemagglutinin component in the complex of 16s progenitor toxin from Clostridium botulinum (HA33) and a ricin-like lectin from Clitocybe nebularis (CNL), which have been developed as a drug-carrier and an anticancer agent, respectively.3,4) In nature, LSMT occurs in its mature form (mLSMT), likely as a result from proteolytic digestion.5) The protein forms a stable complex with PPO36) and their separation has not been successful.
Recently, the full-length recombinant protein using Escherichia coli (rLSMT) that shares an identical structure to mLSMT has been obtained.7) The preliminary study in silico predicted a similarity in biological relevance of LSMT to HA33 or CNL.8) The subsequent study ex vivo unraveled the ability of rLSMT to permeate from a freshly prepared jejunum-derived dialysis bag.9) The protein appears non-toxic and non-immunogenic to inbred and outbred mice upon repeated and extended administration.10,11) This is the basis for LSMT development as a drug carrier as an alternative to the immunogenic HA33.12) The recent characterization of rLSMT demonstrated its affinity towards MCF7 (breast cancer cell), but an effect was observed only at high protein concentrations; therefore, this protein is unattractive for further development as an anticancer agent.5) Finally, rLSMT appears to withstand gastrointestinal track conditions,5) thus it could be employed to deliver drugs of the low intestinal absorptive group via an oral route.
Following that initial premise, we evaluated the development of LSMT as a drug carrier by means of its modification to obtain a bioconjugate under conditions a carrier would meet in vivo. We conjugated rLSMT to captopril, a model of a Biopharmaceutical Classification System (BCS) class III drug. However, the conjugation was not aimed to produce captopril for therapeutic purpose but more to test the capability and capacity of LSMT after being modified. Therefore, this conjugation trial was followed by tests to evaluate the permeability of the conjugate. Captopril was selected because of its reactivity as well as a representative protein for drug delivery with low absorption. First, a structure-based evaluation was performed in silico to outline the strategy to conjugate captopril. rLSMT was chemically modified with a linker to provide the anchor, as a target for captopril. The conjugate was analyzed with TLC and with MS to calculate the amount of captopril bound to rLSMT. The conjugate was tested in vitro using artificial stomach or intestinal fluids to check its stability. Finally, the ability of the conjugate to penetrate the epithelial monolayer barrier of the intestine was evaluated. The rLSMT–captopril conjugate appears to exhibit good stability in the simulated gastrointestinal tract conditions and could migrate across the artificial absorption system of the intestine. The presented results provide a solid basis for our pioneering work to develop LSMT as a drug carrier.
RESULTS AND DISCUSSION
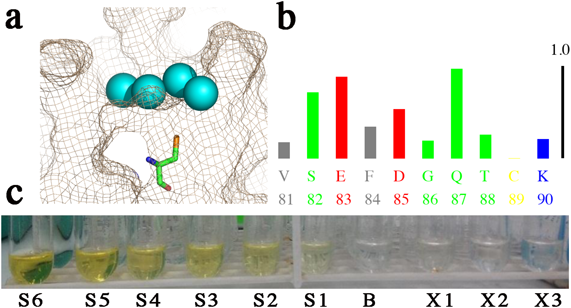
Determining the Conjugation StrategyThe amino acid sequence of rLSMT contains one cystein residue (Cys89) that is located near the protein surface with its thiol side chain pointing towards the surface (Fig. 1a). This situation provides an opportunity for direct interaction between the cysteine with captopril, which also has a free thiol group. However, assessment in silico suggested that the thiol group in the rLSMT is not solvent accessible (Fig. 1b). This prediction was confirmed when no free sulfhydryl group was detected with the Ellman’s reagent (Fig. 1c). A free sulfhydryl group was also not detected at basic pH to increase the reactivity of the cysteine. Hence, the lysine residues of rLSMT were chosen as the new target for modification. Lysine is the most favorite target for chemical modification, because of the reactivity of its primary amine.13) In our case, the modifier reagent should serve as a thiol group to facilitate conjugation of the captopril, therefore the 4-succinimidyloxycarbonyl-alpha-methyl-2-pyridyldithiotoluene (SMPT) cross-linker was used.14) Another reason for using SMPT to link to captopril is that SMTP has previously been used for coupling to lysozyme.15) The pathway for the modification strategy is provided in the supplementary material (Fig. S1).
Captopril–rLSMT ConjugationThe captopril structure contains a free sulfhydryl (thiol) group that contributes to rash and taste impairment in patients. Therefore, alternatives of captopril without a thiol group are popular, such as enalaprilat and lisinopril. Unfortunately, these angiotensin-converting enzyme (ACE) inhibitors also show poor absorption, low bioavailability, and short half-lives.16) On the other hand, captopril is reactive, and can form reversible disulfide bonds with components of the blood, such as albumin, L-cysteine, and glutathione. Thus, captopril appears appealing upon selection of drug model for disulfide bond facilitated conjugation.
Conjugation of captopril to rLSMT was a two-step process: Modification of the protein with the SMPT linker and subsequent conjugation of captopril to the SMPT-modified rLSMT. The TLC analysis during the initial screening (performed in excess of molar ratio of SMPT and captopril to rLSMT) suggested that a conjugate had been formed (Fig. 2). On the TLC plate, free captopril migrated about halfway of the plate while the conjugate did not move. Neither rLSMT nor SMPT-modified rLSMT produced a signal after addition of the 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) visualization reagent.
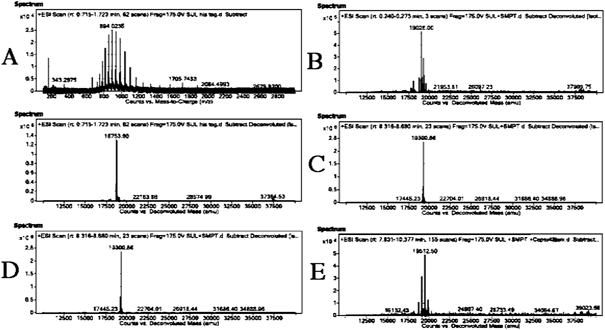
The output from the absolute surface area (ASA)-view program suggested that Lys3 and Lys135 are exposed to the solvent, and are thereby susceptible to modification, while Lys60 and Lys119 are at the 50% cut off. Based on this output, nine possible species of the modified rLSMT were predicted (supplementary material Fig. S1). Following the TLC analysis, MS analysis was performed to identify the conjugate formation based on its intact mass (Fig. 3). De-convoluted mass spectra data from the sample after modification steps is presented in Tables 1 and 2. The intact mass analysis identified the presence of two species after the modification steps. The species were identified as rLSMT with either one or two captopril molecules. No modified rLSMT with a higher conjugation degree was detected. The 1 : 2 : 2 ratio (rLSMT : SMPT : captopril) appeared as the dominant species in the samples, which concurs to the ASA-view that predicted susceptibility of two lysine residues in the protein molecules.
Table 1. Detected Species during Mass Spectrometry Analysis and Their Identity with Masses in Atomic Unit (AU)/Dalton (Da)
| Possible conjugate identified | Mass | Height | Min Z | Max Z | Z Count | Fit score |
|---|
| rLSMT+2 SMPT+2 Captopril | 19512.7739 | 288766 | 9 | 28 | 19 | 8 |
| — | 38993.4318 | 7832 | 30 | 50 | 14 | 8 |
| rLSMT+1 SMPT+1 Captopril | 19133.0723 | 31609 | 15 | 25 | 10 | 8 |
| — | 19495.746 | 21341 | 17 | 24 | 8 | 8 |
| — | 37505.4432 | 2592 | 32 | 48 | 12 | 9 |
| — | 39067.2709 | 1352 | 38 | 48 | 9 | 9 |
Table 2. Tested Conditions to Generate the Conjugate
| Conditions | Mol ratio | Temperature (°C) | Time (h) | Result |
|---|
| 1 | Modification | 1 rLSMT : 4 SMPT | 25 | 2 | Modified rLSMT at 1 : 1 ratio |
| Conjugation | 1 Activated rLSMT : 8 Captopril | 4 | Captopril conjugated rLSMT at 1 : 1 : 1 ratio |
| 2 | Modification | 1 rLSMT : 10 SMPT | 4 | 24 | Modified rLSMT at 1 : 1 and 1 : 2 ratio |
| Conjugation | 1 Activated rLSMT : 100 Captopril | 48 | Captopril conjugated rLSMT at 1 : 1 : 1 and 1 : 2 : 2 ratio |
| 3 | Modification | 1 rLSMT : 10 SMPT | 25 | 24 | No modified rLSMT |
| Conjugation | 1 Activated rLSMT : 100 Captopril | 48 | No conjugate |
The ratio was based on mass spectrometry analysis.
Several conditions were tested, for both modification and conjugation steps, at different molar ratio’s of SMPT linker and captopril, temperature, and contact times (Table 2). A quick reaction performed at room temperature (ca. 25°C) resulted in captopril-conjugated rLSMT at 1 : 1 : 1, thus only one SMPT was bound. Increasing the ratio of the linker to rLSMT resulted in captopril-conjugated rLSMT at 1 : 2 : 2, thus more bound SMPT. This indicates that, of Lys3 and Lys135, one is more susceptible or reactive. The species with 1 : 2 : 2 ratio was obtained when the reaction was carried out at 4°C at a rLSMT : SMPT : captopril ratio of 1 : 10 : 100 at a total reaction time of 72 h.
On one occasion, a conjugate was observed with a molecular mass resembling 1 rLSMT with 3 SMPT molecules, but this result could not be reproduced: Further conjugation tests using the same reaction mixture and conditions produced only an 1 : 2 : 2 conjugate. This suggests that only rLSMT modified by two SMPT molecules were conjugated to captopril. This 1 : 3 species was therefore regarded as an artifact. Interestingly, no product was obtained when the modification and subsequent conjugation reaction was replicated at 25°C rather than 4°C. It is unlikely that this was due to an impaired or unsuccessful reaction, and is probably caused by the instability of the modified rLSMT. The turbidity of the reaction mix increased immediately when SMPT was added to the rLSMT solution. Then, sedimentation occurred over the course of the incubation. This was also observed during the reaction at 4°C, but in that case, the turbidity slowly disappeared and no sediment was formed. Thus, the reaction appears to favor a low temperature (4°C) with a longer incubation time. This is important for future chemical modification of rLSMT that will target the lysine residues. In addition, the amount of captopril required for conjugation to the SMPT-modified rLSMT depends on the number of SMPT linkers attached. However, when the excess amount of captopril was given, the conjugation efficiency decreased due to self-reaction competition.17) In principle, more captopril could be attached, which requires more linker molecules available for binding.
Conjugate StabilityThe stability of captopril-conjugated rLSMT was evaluated by measuring the number of free sulfhydryl groups after two h of incubation of the conjugate in simulated body fluids. The number of free sulfhydryl groups is directly correlated to the number of disrupted disulfide bond in the protein conjugate. As both captopril and SMPT-activated rLSMT react with the DTNB reagent, the number of observed sulfhydryl groups are equivalent with twice disrupted disulfide bonds number. The percentage of intact disulfide bonds in the reaction mixture is presented in Table 3. Previously, LSMT has been shown to be stable against degradation in the gastrointestinal tract environment.5) Further, SMPT protein conjugate is also stable under gastrointestinal conditions as demonstrated in the generation of insulin derivative for oral administration.18) The result suggests that the conjugate is stable in the simulated body and gastrointestinal tract fluids. Therefore, the conjugate is likely also stable in vivo.
Table 3. Stability of the Conjugate in the Simulated Body and Gastrointestinal Tract Fluids
| Medium | Free sulfhydryl (nM) | Intact disulfide bond (%) |
|---|
| Stomach (without digestive enzymes) pH 1.2 | 7.0±0.02 | 94–96 |
| Intestine (without digestive enzymes) pH 6.8 | 6.6±0.01 | 98 |
| Body fluid (physiological) pH 7.2 | 6.5±0.00 | 99 |
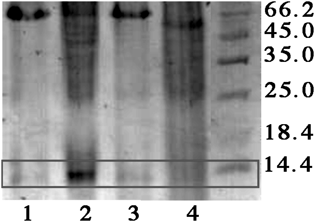
Permeability was measured to establish whether modification interferes with the capability of rLSMT to penetrate the epithelial monolayer barrier of the intestine. The ex vivo testing with a freshly prepared jejunum indicated the presence of both rLSMT and the conjugate in the receptor compartment (Fig. 4). This suggests that the modified rLSMT was able to permeate through the jejunum. The size of modified rLSMT recovered in the dialysis solution was smaller than that of unmodified rLSMT. This is consistent with our previous finding for rLSMT that the protein is fragmented to a species resembling mLSMT.9) Most importantly, this result further suggests that the conjugation caused no significant change of the rLSMT molecule. Hence, the absorbability of rLSMT in the intestine was not altered. In addition, an in vitro assay using a transwell setup was tested (supplementary material Fig. S2). During a sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis, two protein bands were observed where the lower molecular weight protein was dominant. They might represent the rLSMT that was modified by SMPT and by SMPT–captopril because SMPT and captopril are covalently bound to rLSMT. Release of captopril during the transfer was anticipated by a redox system in the intestinal monolayer cells.18) However, these preliminary results indicate that the conjugate remains intact during the transfer through the Caco-2 cells monolayer barrier.
Oral administration is one of the most preferred routes in drug delivery because it offers an easy, safe, and comfortable way of administering drugs. Oral delivery requires no sterility upon administration, and most importantly prevents fear for and injuries by needles in parenteral administration. However, oral drug administration has its own obstacles. The first obstacles are the harsh conditions in the gastrointestinal tract i.e., acidic pH and the presence of digestive enzymes, imposing limitations of the number and type of drugs to deliver, especially for drugs that may suffer from pre-systemic degradation or metabolism.19) The second obstacle is the absorption of a drug into the blood circulation system, especially for drugs with low solubility and/or permeability through the epithelial layer of the gastrointestinal tract. While the issue with gastrointestinal tract has successfully been solved by recent technology for controlled or delayed drug release such as encapsulation or coating of the drug in tablet formulation,20) problems with drug permeability are still not firmly solved. One of the most investigated strategies to improve drug absorption is the use of lectins,21) especially those with low toxicity/immunogenicity and with good display of drug transfer in the intestine.22) In our case, LSMT has shown a promise but proof of concept is required. Our present works showed that rLSMT could be modified safely, without compromising its functionality. Conjugation was also relatively easy and straightforward, but the number of modifiable lysine residues on the protein limits the efficiency of conjugation. This efficiency could be improved by introducing more lysine residues by i.e., site-directed mutagenesis. Other linkers with different functional groups should be looked for to obtain linkers with economic value that would support further development for commercial purposes. rLSMT might extend the number of biomolecules suitable for drug delivery from the BCS III and IV that face problems with low absorption upon oral administration.
CONCLUSION
Modified rLSMT displayed intestinal permeability similar to its native form, while remaining stable in simulated physiological and gastrointestinal fluids. Hence, rLSMT is promising for further development as a drug carrier for oral route of administration.
MATERIALS AND METHODS
MaterialsAll chemicals and reagents were purchased from Sigma (St. Louis, MO, U.S.A.) or Merck (Darmstadt, Germany), except when specifically mentioned otherwise. Captopril was purchased from Zhejiang Huahai Pharma (Zhejiang, China), DTNB (Ellman’s reagent) and SMPT were purchased from Thermo Fisher Scientific (Waltham, MA, U.S.A.). Animal experimentation and protocols were reviewed and approved by the Animal Research Ethics Committee of the Institut Teknologi Bandung Indonesia (Certificate No. 01/KEPHP-ITB/022016).
Production and Purification of rLSMTrLSMT was prepared according to the previous report.9) Briefly, E. coli BL21 (DE3)-rLSMT was grown at 37°C in LB medium containing 100 µg/mL ampicillin for 10–12 h (overnight). Protein expression was induced by the addition of IPTG to a final concentration of 0.05 mM. The bacterial cells were harvested by centrifugation at 5000 ×g, 4°C for 15 min and then washed with phosphate buffer saline (PBS, 137 mM NaCl, 2.7 mM KCl, and 10 mM phosphate buffer, pH 7.4) prior to resuspension in low-salt PBS (27.5 mM NaCl, 0.5 mM KCl, and 10 mM phosphate buffer, pH 7.4). The cells were disrupted with a microfluidizer M-100P (Microfluidics, Westwood, MA, U.S.A.) at 15000 psi for two cycles. The crude extract was collected by centrifugation at 10000×g, 4°C for 15 min and then loaded onto a complete Ni-NTA affinity column (Roche, Singapore) that had been equilibrated with the low-salt PBS buffer. Purified His6-tagged rLSMT was recovered by elution with the same buffer containing 200 mM imidazole. A final polishing purification was done with an EnrichTM SEC70 size exclusion column (BioRad) in low-salt PBS buffer. Purification was performed at 20°C on an NGC Scout Plus purification system (BioRad).
Protein AnalysisThe protein concentration was determined using the Bradford microassay (BioRad) or measurement at 280 nm using the experimentally determined molar coefficient. SDS PAGE analysis was performed using 16% polyacrylamide gel. The protein was visualized with coomassie brilliant blue or silver nitrate staining, depending on protein content or sample conditions.
Screening for Conjugation StrategySolvent accessibility of amino acids in the protein was predicted in silico with the programs ASAView23) and NetSurfP24) using the structure of rLSMT (PDB ID: 5eha) and its amino acid sequence as the input, respectively. Cys89 was targeted for immediate conjugation with captopril. The number of free sulfhydryl groups was determined using Ellman’s reagent DTNB according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA, U.S.A.). Briefly, 20 µL of 4 mg/mL DTNB in 100 mM phosphate buffer, pH 8, containing 1 mM ethylenediaminetetraacetic acid (EDTA), was added onto 1 mL of protein sample in a reaction tube. After 15 min of incubation at room temperature (25°C), the chromogenic TNB compound released was measured at 412 nm with an Agilent Cary 60 spectrophotometer (Agilent Technologies, Singapore). Cysteine hydrochloride solution at concentrations of 0.25–1.5 mM was employed as the standard.
Conjugation of Captopril to LSMTA mixture of SMPT and rLSMT in 100 mM phosphate buffer, pH 7.2, containing 150 mM NaCl, was incubated to obtain the modified protein. Excess of SMPT was removed by buffer exchange over a 3-kDa cut-off amicon membrane (Millipore, Singapore). Next, captopril was added into the SMPT-modified rLSMT in 100 mM phosphate buffer, pH 7.2, containing 150 mM NaCl and 10 mM EDTA to obtain captopril-conjugated rLSMT. The incubation conditions for both processes (conjugation of the linker and of the drug) were varied in temperature, time, and molar ratio. Conditions with higher and consistent yield were adopted to provide sample for bioanalysis of the conjugate.
Analysis of the Protein ConjugateThe modified rLSMT was analyzed with a normal phase thin layer chromatography according to the standardized analytical method for captopril described in the U.S. pharmacopoeia.25) Further analysis was performed with LC-MS to determine the absolute intact mass of the conjugate. The separation was carried out using an LC system (Agilent HPLC 1290 Infinity II) with an Agilent Zorbax Eclipse Plus C18 reversed phase chromatography column (1.8 mm, 2.1×50 mm) thermostated at 40°C. The sample was eluted using 0.1% formic acid (A) and 0.1% formic acid in acetonitrile (B) as the solvents, at a flow rate of 0.4 mL/min with a 10–80% gradient B in 20 min. Data were collected in the positive mode electrospray ionization (ESI) with an MS detector (Agilent 6545 Q-TOF/MS). The capillary, nozzle, and fragmentor voltages were 3500, 1000, and 175 V, respectively. For the MS detector, nitrogen (the drying gas) was heated to 320°C and injected at the rate of 8 L/min. Additional heating was applied using sheath gas heated to 350°C and at flow rate of 11 L/min. The data was processed with the Agilent Mass HunterTM Qualitative Analysis program (ver. B.07). Maximum entropy deconvolution algorithm was applied to determine the absolute intact mass of the conjugate. Selected mass range for deconvolution result analysis was in the range of m/z 10000–40000 at 1 Da mass steps. The output was refined by imposing the baseline factor of 7.0 and by the choosing of only the proton adduct.
Stability of the conjugate was tested in simulated intestine and body fluid.26,27) Briefly, a known amount of modified rLSMT was mixed with the designated simulation fluid at ratio 1 : 1. The mixture was then incubated at 37°C. The increase in the number of sulfhydryl groups detected in the fluid after 1 h of incubation was determined with Ellman’s reagent.
Permeability StudyThe permeability study was performed by the non-everted gut sac method as previously reported.9) First, a Wistar rat was acclimatized without food but with free access to drink water for 12 h prior to sacrificing it in a CO2 chamber (3–4 min at 30% CO2). The jejunum was collected and immediately divided into three of five-centimeter segments. Each segment was washed with cold PBS and one of its ends was carefully tied with surgical suture. Next, the sample (modified or unmodified rLSMT) was inserted into jejunum that its open end was then carefully tied with surgical suture. This jejunum containing sample was placed in a beaker glass containing 20 mL PBS (dialysis buffer) with the two ends not immersed in the solution (U-shape) and then incubated at 37°C, 50 RPM for 2 h on an orbital shaker (Infors AG, Bottmingen, Switzerland). Afterwards, the solution in the jejunum (donor) and the dialysis buffer (acceptor) were independently collected and concentrated with TCA prior to SDS-PAGE analysis (Mini-protean system, BioRad).
Acknowledgment
The works were funded by the Ministry of Research and Higher Education coordinated by the Higher Education Excellent Research through the Program Unggulan Perguruan Tinggi (PUPT, Contract No. 127/SP2H/PTNBH/DRPM/2018) and supported by Dexa Medica. We thank Dr. H. J. Doddema for helping with the writing and language editing.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Ismaya WT, Rozeboom HJ, Weijn A, Mes JJ, Fusetti F, Wichers HJ, Dijkstra BW. Crystal structure of Agaricus bisporus mushroom tyrosinase: Identity of the tetramer subunits and interaction with tropolone. Biochemistry, 50, 5477–5486 (2011).
- 2) Weijn A, Bastiaan-Net S, Wichers HJ, Mes JJ. Melanin biosynthesis pathway in Agaricus bisporus mushrooms. Fungal Genet. Biol., 55, 42–53 (2013).
- 3) Ito H, Sagane Y, Miyata K, Inui K, Matsuo T, Horiuchi R, Ikeda T, Suzuki T, Hasegawa K, Kouguchi H, Oguma K, Niwa K, Ohyama T, Watanabe T. Ha-33 facilitates transport of the serotype d botulinum toxin across a rat intestinal epithelial cell monolayer. FEMS Immunol. Med. Microbiol., 61, 323–331 (2011).
- 4) Pohleven J, Obermajer N, Sabotič J, Anžlovar S, Sepčić K, Kos J, Kralj B, Štrukelj B, Brzin J. Purification, characterization, and cloning of a ricin b-like lectin from mushroom Clitocybe nebularis with antiproliferative activity against human leukemic t cells. Biochim. Biophys. Acta, 1790, 173–181 (2009).
- 5) Ismaya WT, Tandrasasmita OM, Sundari S, Diana, Lai X, Retnoningrum DS, Dijkstra BW, Tjandrawinata RR, Rachmawati H. The light subunit of mushroom Agaricus bisporus tyrosinase: Its biological characteristics and implications. Int. J. Biol. Macromol., 102, 308–314 (2017).
- 6) Ismaya WT, Rozeboom HJ, Schurink M, Boeriu CG, Wichers H, Dijkstra BW. Crystallization and preliminary X-ray crystallographic analysis of tyrosinase from the mushroom Agaricus bisporus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun., 67, 575–578 (2011).
- 7) Lai X, Soler-Lopez M, Ismaya WT, Wichers HJ, Dijkstra BW. Crystal structure of recombinant tyrosinase-binding protein MtaL at 1.35 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun., 72, 244–250 (2016).
- 8) Ismaya WT, Yunita, Damayanti S, Wijaya C, Tjandrawinata RR, Retnoningrum DS, Rachmawati H. In silico study to develop a lectin-like protein from mushroom Agaricus bisporus for pharmaceutical application. Sci. Pharm., 84, 203–217 (2016).
- 9) Ismaya WT, Yunita, Efthyani A, Lai X, Retnoningrum DS, Rachmawati H, Dijkstra BW, Tjandrawinata RR. A novel immune-tolerable and permeable lectin-like protein from mushroom Agaricus bisporus. Biochem. Biophys. Res. Commun., 473, 1090–1093 (2016).
- 10) Ismaya WT, Efthyani A, Retnoningrum D, Lai X, Dijkstra B, Tjandrawinata R, Rachmawati H. Study of response of swiss webster mice to light subunit of mushroom tyrosinase. Biotech. Histochem., 92, 411–416 (2017).
- 11) Ismaya WT, Efthyani A, Tjandrawinata RR, Rachmawati H. Biological responses in balb/c mice after long-term parenteral administration of the light subunit of mushroom tyrosinase. J. Biochem. Mol. Toxicol., 31, e21958 (2017).
- 12) Sayadmanesh A, Ebrahimi F, Hajizade A, Rostamian M, Keshavarz H. Expression and purification of neurotoxin-associated protein ha-33/a from Clostridium botulinum and evaluation of its antigenicity. Iran. Biomed. J., 17, 165–170 (2013).
- 13) Boutureira O, Bernardes GJL. Advances in chemical protein modification. Chem. Rev., 115, 2174–2195 (2015).
- 14) Méndez TJ, Johnson JV, Nichols LS, Lang GHL, Eyler JR, Powell DH, Richardson DE. Bioconjugation of ribonuclease a: A detailed chromatographic and mass spectrometric analysis of chemical modification by a cross-linking reagent. Bioconjug. Chem., 11, 182–194 (2000).
- 15) Kok RJ, Grijpstra F, Walthuis RB, Moolenaar F, de Zeeuw D, Meijer DK. Zeeuw Dd, Meijer DKF. Specific delivery of captopril to the kidney with the prodrug captopril-lysozyme. J. Pharmacol. Exp. Ther., 288, 281–285 (1999).
- 16) Kelly JG, O’Malley K. Clinical pharmacokinetics of the newer ace inhibitors. A review. Clin. Pharmacokinet., 19, 177–196 (1990).
- 17) Bojarska J, Maniukiewicz W, Fruziński A, Sieroń L, Remko M. Captopril and its dimer captopril disulfide: Comparative structural and conformational studies. Acta Crystallogr. C, 71, 199–203 (2015).
- 18) Circu ML, Aw TY. Redox biology of the intestine. Free Radic. Res., 45, 1245–1266 (2011).
- 19) Pereira de Sousa I, Bernkop-Schnürch A. Pre-systemic metabolism of orally administered drugs and strategies to overcome it. J. Control. Release, 192, 301–309 (2014).
- 20) Andrade C. Sustained-release, extended-release, and other time-release formulations in neuropsychiatry. J. Clin. Psychiatry, 76, e995–e999 (2015).
- 21) Varrot A, Basheer SM, Imberty A. Fungal lectins: Structure, function and potential applications. Curr. Opin. Struct. Biol., 23, 678–685 (2013).
- 22) Woodley JF. Lectins for gastrointestinal targeting – 15 years on. J. Drug Target., 7, 325–333 (1999).
- 23) Ahmad S, Gromiha M, Fawareh H, Sarai A. Asaview: Database and tool for solvent accessibility representation in proteins. BMC Bioinformatics, 5, 51 (2004).
- 24) Petersen B, Petersen TN, Andersen P, Nielsen M, Lundegaard C. A generic method for assignment of reliability scores applied to solvent accessibility predictions. BMC Struct. Biol., 9, 51 (2009).
- 25) World Health Organization, Essential medicines and pharmaceutical policies 6th (2016).
- 26) Marques MRC, Loebenberg R, Almukainzi M. Simulated biological fluids with possible application in dissolution testing. Dissolut. Technol, 18, 15–28 (2011).
- 27) United States of America Pharmacopoeia, National formulatory, 30 (2012).