3. RECENT ADVANCES IN OLIGONUCLEOTIDE-BASED THERAPY IN ATTR-FAP
On the basis of the current most effective therapies for ATTR-FAP, there is growing evidence that reduction of TTR production by the liver is a promising therapeutic approach for the treatment of ATTR-FAP.21–24) Based on the above theoretical rationale, a silencing approach of TTR by oligonucleotide-based drugs has been widely developed, with clinical studies now underway (Table 2).
Table 2. Clinical and Preclinical Development of an Oligonucleotide-Based Therapeutic Approach for ATTR-FAP
| Oligonucleotide type | Name | Delivery system | Administration | Status | Reference |
|---|
| Small interfering RNA (siRNA) |
| siRNA | Patisiran (ALN-TTR02) | LNP (lipid-based delivery) | Intravenous | Phase III (complete) | 39–41) |
| Revusiran (ALN-TTRsc) | GalNAc-siRNA | Subcutaneous | Phase III (discontinue) | 42–46, 48) |
| ALN-TTRsc02 | GalNAc-ESC-siRNA | Subcutaneous | Preclinical | 47, 49) |
| Mutant TTR selective siRNA | — | (in vitro, COS-7 cell) | — | 50) |
| Lac-α-CDE/siRNA complex | CDE complex (polymer-based delivery) | Intravenous | — | 51, 52) |
| Antisense oligonucleotide (ASO) |
| ASO | Inotersen (IONIS-TTRRx) | Chemically modified ASO | Subcutaneous | Phase III (complete) | 72–74) |
| AKCEA-TTR-LRx (IONIS-TTR-LRx) | GalNAc-ASO | Subcutaneous | Preclinical | 75, 76) |
| DNA/RNA duplex | HDO | Toc-HDO (LNA/DNA/LNA gaper) | Intravenous | — | 77) |
| Genome editing |
| SSOs | SSOs/AC complex | SSOs/AC complex | Hepatocyte injection | — | 78) |
| CRISPR/Cas system | CRISPR/Cas9 LNP | LNP | Intravenous | — | 89) |
LNP: lipid nanoparticle, GalNAc: N-acetylgalactosamine, ESC: enhanced stability chemistry, Lac-α-CDE: lactosylated dendrimer/cyclodextrin conjugate, Toc: α-tocopherol, LNA: locked nucleic acid, SSOs: single stranded oligonucleotides, AC: atelocollagen.
RNA interference (RNAi) is a well understood mechanism in mammalian cells by which sequence-specific RNA molecules, such as siRNA, miRNA or shRNA, lead to targeting and cleaving complementary mRNA.25) The discovery of RNAi has had a great impact on the development of novel therapeutic strategies for intractable diseases such as cancer,26,27) viral infections,28,29) and hereditary disorders.3,30) In particular, siRNA-based therapy has demonstrated promising outcomes in clinical trials.3,31–33) In order to realize the optimum effects of siRNA-based therapy, efficient in vivo siRNA delivery into the cytoplasm of target cells in target organs is required. However, due to siRNA’s distinct physicochemical and biological properties, such as relatively large molecular weight, hydrophilicity, anionic charge, instability in the bloodstream, immune response activation and rapid clearance,3,34,35) various hurdles stand in the way of effective in vivo siRNA delivery.
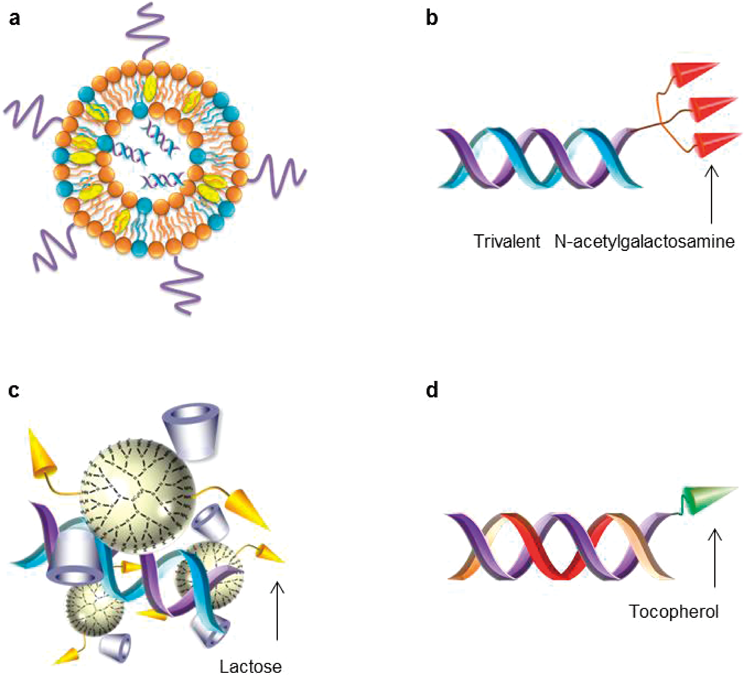
Alnylam Pharmaceuticals has developed several siRNA-based drugs for the treatment of ATTR-FAP. These siRNA drugs (TTR-targeted siRNA) can suppress TTR expression, since they specifically bind to the 3′-untranslated region (3′-UTR) of human TTR mRNA.36) This company also developed novel TTR-targeted siRNA encapsulated lipid nanoparticles (LNPs), called ALN-TTR01 and ALN-TTR02, which are first- and second-generation formulations of LNPs (Fig. 2a). Although these LNPs have almost identical physicochemical properties, components of their functional lipids are extremely different, a key factor in their in vivo RNAi effect.37) After intravenous administration, the LNPs are opsonized by apolipoprotein E (ApoE) and then enter the liver from blood circulation through endothelial fenestrae, where they bind to low density lipoprotein receptors (LDLR) on the hepatocyte surface. The LNPs are taken up via LDLR-mediated endocytosis, then protonation of the ionizable lipid component of the LNP occurs, due to the acidic pH of the endosome. The positively charged LNP fuses with the negatively charged endosomal membrane, leading to the release of siRNA into the cytosol and subsequent loading onto the RNA-induced silencing complex (RISC). Finally, argonaute protein (Ago2) within RISC then mediates TTR mRNA cleavage, leading to the knockdown of TTR expression.38) In non-human primates (cynomolgus monkeys), ALN-TTR01 treatment reduced TTR expression by up to 50% at day 7, at a dose of 1.0 mg/kg. ALN-TTR02 also showed significant improvement of knockdown efficacy. ALN-TTR02 treatment reduced TTR expression by up to 75% at day 7, at a dose of 0.1 mg/kg, and by approximately 90% at day 14, at a dose of 0.3 mg/kg. Furthermore, approximately 70% suppression of serum TTR was maintained at day 28 at a dose of 0.3 mg/kg.39)
In a phase 1 study, ALN-TTR01 suppressed TTR, with a mean reduction of about 40% at day 7, at a single dose of 1.0 mg/kg, as compared with a placebo. Additionally, ALN-TTR02 suppressed TTR with a mean reduction of 80% at day 7, at a dose of 0.3 mg/kg, with a sustained reduction of more than 60% at 28 d. ALN-TTR01 and ALN-TTR02 produced 21 and 8% mild-to-moderate adverse events (AE), respectively. Based on these clinical trial results, ALN-TTR02 has performed as patisiran.39) In a phase 2 study, ATTR-FAP patients were administered intravenous infusions of patisiran every 3 or 4 weeks in a dose range of 0.01–0.3.40) The administration of patisiran induced rapid, dose-dependent, and persistent TTR suppression, with 0.3 mg/kg patisiran treatment exhibiting the maximum effect. In the patisiran group, mild-to-moderate AEs occurred in 10% of patients (infusion-related reactions), and 4 serious AEs (SAEs) were reported (urinary tract infection, sepsis, nausea/vomiting, extravasation-related cellulitis). Recently, Adams et al. reported the results of a phase 3 APOLLO study, a multicenter, multinational, randomized, double-blind, placebo-controlled, to evaluate the efficacy and safety of patisiran in ATTR-FAP patients with polyneuropathy.36,41) In the APOLLO study, they evaluated various clinical scores of ATTR-FAP, such as the modified Neuropathy Impairment Score (mNIS), Norfolk QOL-Diabetic Neuropathy (QOL-DN), mBMI, and so on. The difference in the least-squares mean change from baseline at 18 months between the two groups (patisiran minus placebo) was −34.0 points for the mNIS+7 and −21.1 points for the Norfolk QOL-DN score. Patisiran also ameliorated the gait speed and mBMI. The difference in least-squares mean change from baseline between patisiran and the placebo was 0.31 m/s for gait speed, and 115.7 for mBMI at 18 months. On the whole, 97% of the patients reported mild-to-moderate AEs, and the frequency of SAEs was similar in the two groups (36% in the patisiran group and 40% in the placebo group). The APOLLO study demonstrated that patisiran improved multiple clinical manifestations and provided benefits to ATTR-FAP patients. Most recently, ONPATTRO™ (trade name of patisiran) was approved by the FDA (August 10, 2018) as a first siRNA-based drug.
ALN-TTRsc (Revusiran), another type of siRNA-based drug, had also been in development as an ATTR-FAP therapeutics (Fig. 2b). Revusiran is composed of a 2′-O-methyl and 2′-deoxy-2′-fluoro modified siRNA conjugated with triantennary N-acetylgalactosamine (GalNAc).42) GalNAc residue specifically associates with the asialoglycoprotein receptor (ASGPR), which is highly expressed on hepatic parenchymal cells, thus revusiran is taken up via ASGPR-mediated endocytosis, and suppresses TTR expression in hepatocytes.43,44) In preclinical studies, revusiran induced hepatocyte-specific, persisting, and potent TTR knockdown in mice and non-human primate models.42,45)
In the phase 1 study, subjects were administered a subcutaneous injection of revusiran at a dose range of 1.25–10 mg/kg in single or multiple ascending dose phases.43) The major common AE of revusiran was temporary mild-to-moderate injection site reaction. Revusiran significantly reduced serum TTR level compared to the placebo, with a mean maximum reduction of approximately 90% at a dose of 2.5–10 mg/kg. These results demonstrate that the GalNAc conjugated siRNA (GalNAc-siRNA) platform holds enormous potential for liver-based disease treatment. A phase 2 and open label extension demonstrated a more than 90% reduction in serum TTR level after multiple dosing, with a persistent knockdown of TTR beyond 90 d in ATTR-FAP patients with cardiomyopathy. Following a 12-month treatment period, five of nine patients had met the primary endpoint goal of being able to take a stable 6 min walk.46,47) However, in an ENDEAVOUR phase 3 study, revusiran development was discontinued due to an imbalance in mortality: 16 deaths in the revusiran group compared with 2 deaths in the placebo group.47–49)
Recently, a novel GalNAc-siRN called ALN-TTRsc02, which is an enhanced stabilization chemistry (ESC)-siRNA-GalNAc conjugate, has been developed.47,49) In the phase 1 study, ALN-TTRsc02 elicited persisting and potent serum TTR suppression, with a mean maximum reduction of 97%, and more than 80% TTR knockdown after nearly 1 year at a single 50 mg dose. The required administration amount of ALN-TTRsc02 is much lower than that of revusiran due to novel ESC technology. Additionally, ALN-TTRsc02 is generally well tolerated, with mild-to-moderate AEs and no SAEs. ALN-TTRsc02 will enter its phase 3 study in late 2018, with an anticipated further improvement in dosing and safety profile for ATTR-FAP patients.
Several other groups have reported other TTR gene modifying therapeutic approaches using siRNA. Kurosawa et al. designed Val30Met TTR selective silencing siRNA in vitro.50) Selective siRNA were designed to target the Val30Met TTR coding region, including the point mutation, which is a different target region from Alnylam’s siRNA. When used in wild-type and Val30Met TTR expression vector transfected COS-7 cells, these siRNA selectively suppressed Val30Met expression, but not the wild-type. A polymer-based TTR-target siRNA delivery system, called lactosylated dendrimer/cyclodextrin conjugate (Lac-α-CDE), for therapeutic use against ATTR-FAP has also been reported51,52) (Fig. 2c). Lac-α-CDE is efficiently recognized by ASGPR due to a modification of lactose. Lac-α-CDE/TTR-target siRNA complexes elicited prominent RNAi effects with efficient ASGPR-mediated cellular uptake, enhancing endosomal escaping ability and low cytotoxicity in HepG2 cells. Moreover, approximately 30% TTR knockdown and negligible side effects were observed after tail vein injection of the Lac-α-CDE complex in Balb/c mice. Recently, this research group also reported an advanced polymer-based TTR-target siRNA delivery system, called Lac-α-CDE/anionic polysaccharide ternary complex system.53) This Lac-α-CDE/TTR-target siRNA/anionic polysaccharide ternary complex elicited prominent in vivo RNAi effects at a lower dose of siRNA than Lac-α-CDE/TTR-target siRNA complex in hepatocytes of Balb/c mice.
3.2. ASOsASOs are composed of single-strand, typically with 16–20 nucleotides, and synthetic oligonucleotides which associate with target RNA sequences via Watson–Crick hybridization.54) Once bound to target RNA, ASOs affect the target RNA metabolism through a variety of mechanisms, such as endogenous ribonuclease (RNase) H1 mediated degradation,55) leading to target mRNA and protein suppression.56,57) So far, several ASO-based drugs have been approved, and have demonstrated great therapeutic efficacy in cytomegalovirus (CMV) retinitis,58–60) homozygous familial hypercholesterolemia,61–64) Duchenne muscular dystrophy (DMD),65–67) and spinal muscular atrophy (SMA)68–71) (Table 1).
Ionis Pharmaceuticals has developed ASO drugs for the treatment of ATTR-FAP. Inotersen (formerly ISIS-TTRRx, IONIS-TTRRx) is a second-generation 2′-O-methoxyethyl modified ASO, which binds to the 3′-UTR of human TTR, resulting in the suppression of TTR expression. In a preclinical study involving a human TTR transgenic mouse and a non-human primate model, inotersen treatment reduced hepatic and serum TTR expression by up to 80%.72,73)
In the phase 1 study, after subcutaneous administration, inotersen showed 96% serum TTR suppression without any AEs in healthy volunteers.73) Recently, Benson et al. reported the result of a phase 3 NEURO-TTR study to demonstrate the safety and efficacy of inotersen treatment in ATTR-FAP patients, including in the presence of cardiomyopathy.74) The difference in the least-squares mean change from baseline at 66 weeks, between inotersen and the placebo, was −19.7 points for the mNIS+7 and −11.7 points for the Norfolk QOL-DN score. In addition, improvement in the clinical score was independent of disease stage, type of TTR mutation, and the presence of cardiomyopathy. However, 5 deaths were observed in the inotersen group. After treatment with inotersen, the major common SAEs were glomerulonephritis and thrombocytopenia. A NEURO-TTR study demonstrated that inotersen improved clinical manifestations and the course of neurologic disease of ATTR-FAP patients. Now, TEGSEDI™ (trade name of inotersen) is under review by the FDA as a therapeutic for ATTR-FAP.
Furthermore, Akcea Therapeutics and Ionis Pharmaceuticals are co-developing AKCEA-TTR-LRx (formerly IONIS-TTR-LRx) to suppress TTR production from hepatocytes, the same ASOs sequence design as inotersen.2,75) AKCEA-TTR-LRx is a triantennary GalNAc conjugated ASO (GalNAc-ASOs). GalNAc-ASOs showed approximately 10-fold higher TTR suppression than non-conjugated ASOs in human Ile84Ser TTR transgenic mice.76) AKCEA-TTR-LRx will enter clinical trials in 2018 for the treatment of ATTR-FAP.
Nishina et al. have reported a third class of oligonucleotides, DNA/RNA heteroduplex oligonucleotides (HDO).77) HDO are comprised of a main locked nucleic acid (LNA)/DNA/LNA gapmer strand and its complementary RNA (cRNA) strand, which is a different structure from siRNA and ASOs. For hepatocyte-targeted delivery, a cRNA strand was conjugated with α-tocopherol (Toc-HDO, Fig. 2d), which is recognized by the LDLR of hepatocytes. After cellular uptake into cells, endogenous RNase H recognizes the center gap portion of DNA/RNA and cleaves an α-tocopherol conjugated cRNA strand, creating the active parent ASO strand (main LNA/DNA/LNA strand). Then, the ASO strand is hybridized with and cleaves the target RNA via an RNase H mediated mechanism, leading to the suppression of target protein expression. Toc-HDO showed greater potency than the parent single-stranded gapmer ASOs in hepatocytes. After tail vein administration to human Val30Met TTR transgenic mice, Toc-HDO significantly suppressed the hepatic TTR expression compared to the parent ASO: approximately 90% suppression was observed at a dose of 0.75 mg/kg.77) Therefore, HDO technology is expected to be a next generation candidate in the development of ASO-based drugs.
3.3. Genome Editing Therapeutic ApproachIt is expected that the correction of the single mutation of mutant TTR genes will lead to improving tetrameric TTR stability. Nakamura et al. reported a targeted gene repair strategy using single-stranded oligonucleotides (SSOs).78) SSOs were developed in the field of cell-free assays to characterize and improve chimeric RNA/DNA oligonucleotides.79,80) The gene repair efficacy of SSOs is higher than that of chimeric RNA/DNA oligonucleotides, both in cell-free extracts80) and in a yeast system.81) As SSOs are easy to synthesize, making them less expensive compared with chimeric RNA/DNA oligonucleotides, they have great potential for therapeutic use in the treatment of various genetic disorders. And in fact, after SSOs/atelocollagen complex treatment in human Val30Met transgenic mice, approximately 10% of Val30Met TTR was converted to wild-type TTR in the liver.78) Gene repair therapy via SSOs has potential as an alternative to LT in the treatment of ATTR-FAP.
As the function of TTR seems dispensable in vivo, it is expected that persistent knockdown or knockout of TTR production will not lead to severe adverse effects. During the past decade, genome editing technology has developed rapidly, for example, zinc-finger nucleases (ZFNs),82) transcription activator-like effector nucleases (TALENs),83,84) and the CRISPR/Cas system.85) These tools are used to induce double-strand breaks for genome DNA, and the subsequent repair systems could introduce mutations to a targeted site. Therefore, these tools have great potential as therapeutics in the treatment of hereditary disorders.86–88) In particular, the CRISPR/Cas system is known to be relatively more flexible and effective than other genome editing tools. Recently, Finn et al. have reported the utility of LNP-mediated delivery of a TTR-targeted CRISPR/Cas9 system.89) With a single intravenous administration to CD-1 mice and Sprague Dawley rats, the LNP-TTR-CRISPR/Cas system induced significant TTR gene editing in hepatocytes: approximately 70% editing at a dose of 3 mg/kg. Furthermore, approximately 97% of serum TTR suppression persisted for at least 12 months. These results suggest that the CRISPR/Cas system may have great potential as a genome editing based ATTR-FAP therapy.