Abstract
Ginsenoside-Rg1 (G-Rg1) is an agent isolated from Panax ginseng that exerts anti-fibrotic effects; however, the mechanism is still unclear. Herein, we investigated whether G-Rg1 administration can mitigate or reverse unilateral ureteral obstruction (UUO)-induced renal fibrosis by regulating the Klotho/transforming growth factor (TGF)-β1/Smad signaling pathway in rats. Sprague–Dawley male rats were subjected to UUO, and rats in the treatment group were administered G-Rg1 or G-Rg1 plus Klotho short hairpin RNA interference (shRNA), while rats in the control and model groups were administered vehicle for 14 d. Epithelial–mesenchymal transition (EMT) biomarkers and Klotho/TGF-β1 signaling molecules were examined by immunohistochemistry, quantitative real-time PCR and Western blotting. Immunohistochemistry showed that UUO induced increased pro-fibrotic TGF-β1 expression, overexpression of the mesenchymal marker, α-smooth muscle actin (α-SMA), and suppression of the epithelial marker, E-cadherin. Moreover, Western blotting analysis indicated that UUO promoted TGF-β1 and phosphorylated Smad3 (p-Smad3) expression (p<0.01), but blocked Klotho and Smad7 expression (p<0.01). After G-Rg1 administration, the UUO-induced TGF-β1 and p-Smad3 expression was suppressed (p<0.01), whereas the reduced Klotho and Smad7 expression was reversed (p<0.05), followed by amelioration of the EMT process. Intriguingly, the G-Rg1 effects were largely abrogated by Klotho knockdown. Furthermore, Klotho expression was upregulated by G-Rg1 treatment at the mRNA and protein levels. Our results suggest that G-Rg1 may be beneficial for ameliorating renal fibrosis by targeting Klotho/TGF-β1/Smad signaling in UUO rats.
Tubulointerstitial fibrosis (TIF) is a feature of end-stage renal disease (ESRD) that is characterized by inflammatory cell infiltration, epithelial cell dysfunction and myofibroblast activation. One possible mechanism underlying TIF is epithelial-mesenchymal transition (EMT) of tubular epithelial cells.1,2) Transforming growth factor β1 (TGF-β1) and its downstream signaling molecules play a vital role in initiating the EMT process. During EMT, tubular epithelial cells lose epithelial-specific markers, such as E-cadherin, and gain mesenchymal markers, such as fibroblast-specific protein 1 (FSP1) and α-smooth muscle actin (α-SMA). Although the contribution of EMT to TIF has been recently debated, it is generally accepted that EMT of renal tubular epithelial cells occurs in various fibrotic kidney diseases, including diabetic,3) immunoglobulin A4) and obstructive5) nephropathies.
Klotho, which was discovered in 1997, is an anti-aging gene that encodes the Klotho protein.6) Klotho is a multifunctional protein that regulates phosphate metabolism, resistance to reactive oxygen species and lifespan extension. Klotho is abundantly expressed in the kidney, thus it is not surprising that kidney disease reduces its expression.7,8) In vivo and in vitro studies have shown that Klotho deficiency facilitates TIF progression, and experimental models that overexpress Klotho are resistant to renal fibrosis.9,10) Consistent with these observations, we have also shown that Klotho-based therapy ameliorates endoplasmic reticulum (ER) stress and TIF in obstructive nephropathy,11) and suppresses the renal EMT process in chronic cyclosporine nephropathy,12) indicating that Klotho may function as an antifibrotic protein; thus targeting Klotho may have therapeutic potential against TIF.13) It has been reported that TGF-β1-induced fibrotic changes are accompanied by Klotho downregulation.14) Furthermore, it has been reported that low Klotho expression strengthens TGF-β1 activity.10) Klotho has also been proven to be an endogenous antagonist of TGF-β1 signaling activity by directly binding to type-II TGF-β1 receptor and negatively regulating TGF-β1 and its downstream signaling.15)
Ginsenoside Rg1 (G-Rg1) is isolated and purified from Panax ginseng, and is presumably the major active ingredient in this plant, which also possesses various pharmacologic actions.16,17) G-Rg1 is widely used for long for treatment of metabolism disorders, cancers, and cardiovascular and hepatic diseases in oriental countries, especially in China. We have recently reported that G-Rg1 ameliorated cell apoptosis in renal tubular cells, and suppressed TIF through ER stress in cyclosporine A treated rats18) and in unilateral ureteral obstruction (UUO) rats.19) The pharmacological activities of G-Rg1 overlap the functions of Klotho, based on the antifibrotic property, indicating that the effects of G-Rg1 on TIF may be associated with Klotho in the kidney. Continuing our previous study, we established a rat renal fibrosis model by UUO, to investigate the hypothesis that G-Rg1 treatment attenuates renal fibrosis by regulating the Klotho/TGF-β1/Smad signaling pathway and the related EMT process.
MATERIALS AND METHODS
Animals and the UUO Rat ModelTwenty-four Sprague-Dawley male rats (200–250 g) were purchased from Shanghai Sipper-BK laboratory animal Co., Ltd. (Shanghai, China) and randomly divided into four groups (n=6 per group): 1) sham group, 2) model group, 3) G-Rg1-treatment group and 4) G-Rg1 plus Klotho-knockdown group. The UUO rat model was established as previously described.11) G-Rg1 (68317; Sigma-Aldrich, St. Louis, MO, U.S.A.) was administered to the rats by intraperitoneal injection at a dose of 50 mg/kg per day based on our previous results18,19) (in our preliminary experiments, 25 mg/kg failed to yield enough beneficial effects and 100 mg/kg failed to yield extra beneficial effects). Klotho knockdown was mediated by short hairpin RNA interference (shRNA). Rats received a single administration of plasmid DNA (50 ng) containing the klotho shRNA (shKl) sequence or a scramble shRNA (shNC) (OriGene Technologies, Rockville, MD, U.S.A.)20) by intravenous injection one day before the UUO operation. Rats in the other groups were injected with physiological saline. All animal experiments were approved by the Animal Care and Use Committee of the Kunshan First People’s Hospital. Fourteen days after the UUO operation, all rats were anesthetized and sacrificed, and then kidney specimens were removed and harvested on the same day. The collected samples were processed for histopathology, immunohistochemistry, quantitative real-time PCR and Western blotting analysis.
Measurement of Biochemical IndicatorsBody weight was monitored every day and whole blood was withdrawn on day 14. The concentrations of serum creatinine (Scr) and blood urea nitrogen (BUN) were determined using an automatic biochemical analyzer.
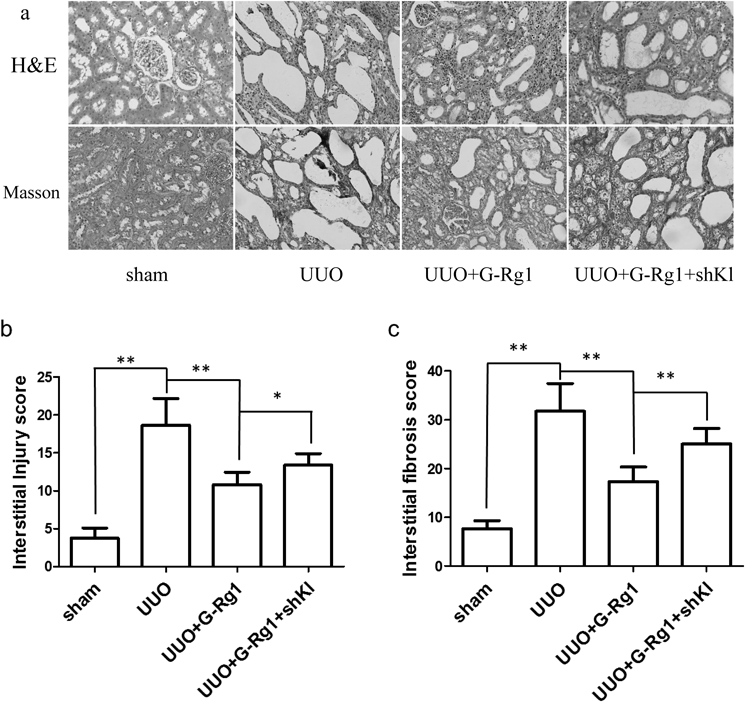
Renal Interstitial Fibrosis and Semi-quantitative Analysis of Renal Injury IndexKidney tissues were sectioned into small blocks and fixed with 10% formalin. After fixation and dehydration, samples were embedded in paraffin and cut into 4-µm-thick sections, which were stained with hematoxylin and eosin (H&E), to assess the interstitial injury severity. Parameters including cell infiltration, edema, tubular atrophy and interstitial fibrosis were examined and scored from 0 to 4, based on a previous study.11) Masson’s trichrome staining was performed to evaluate the interstitial fibrosis severity by scanning 10 non-repeated fields. The extent of interstitial fibrosis was quantified using Image Pro plus-6.0 software and the results are presented as the proportion of fibrotic areas in the scanned interstitial area.
RNA Isolation and Quantitative (q)PCRTotal RNA from kidney tissues was isolated with TRIzol reagent (Ambion Life Tech., U.S.A.). Next, 1 µg of total RNA was used to prepare cDNA using HiScript II Q Select SuperMix for qPCR (R232-01; Vazyme Biotech, U.S.A.) and an ABI GeneAmp 9700 PCR System (Applied Biosystems, U.S.A.). Subsequently, qPCR analysis was carried out using ChamQ SYBR qPCR Master Mix (Q311-02; Vazyme Biotech, U.S.A.) with an ABI 7500 Fast Real-Time PCR System (Applied Biosystems), according to the manufacturer’s instructions. The relative level of Klotho was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and examined using the 2–ΔΔCT method. The primers used were: Klotho, forward 5′-CGT GAA TGA GGC TCT GAA AGC-3′, reverse 5′-GAG CGG TCA CTA AGC GAA TAC G-3′; GAPDH, forward 5′-ATG GGA AGC TGG TCA TCA AC-3′, reverse 5′-CCA CAG TCT TCT GAG TGG CA-3′.
ImmunohistochemistryImmunohistochemistry was performed on 4-µm-thickn sections following dewaxing and dehydration. Processed specimens were immunostained with primary antibodies against TGF-β1 (NBP1-67698; NOVUS,), α-SMA (A5228; Sigma-Aldrich) and E-cadherin (ab1416; Abcam). Immunostaining was performed according to the manufacturers’ instructions. Stained sections were examined and photographed under light microscopy at 200× magnification.
Western BlottingKidney tissues were processed and immunoblot analyses were conducted using the following specific primary antibodies: Klotho Rabbit antibody (Ab) (ab203576; Abcam), TGF-β1 Rabbit Ab (ab92486; Abcam), Phospho-Smad3 (Ser423/425) (C25A9) Rabbit monoclonal antibody (mAb) (#9520; CST, Danvers, MA, U.S.A.), Smad7 Rabbit mAb (#293039; R&D) and GAPDH Rabbit mAb (#5174; CST), and anti-rabbit secondary antibodies (Beyotime, A0208). Protein expression was quantified by normalizing the protein of interest to GAPDH. The ChemStudio system (Analytik Jena AG) was used to detect protein signals, and AJ VisionWorks software was used for densitometric analyses.
Statistical AnalysisAll data are presented as mean±standard deviation (S.D.). Multiple comparisons among groups were determined by a one-way ANOVA followed by Student–Newman–Keuls test. p values less than 0.05 were considered statistically significant. All statistical analyses were conducted using SPSS 19.0 software.
RESULTS
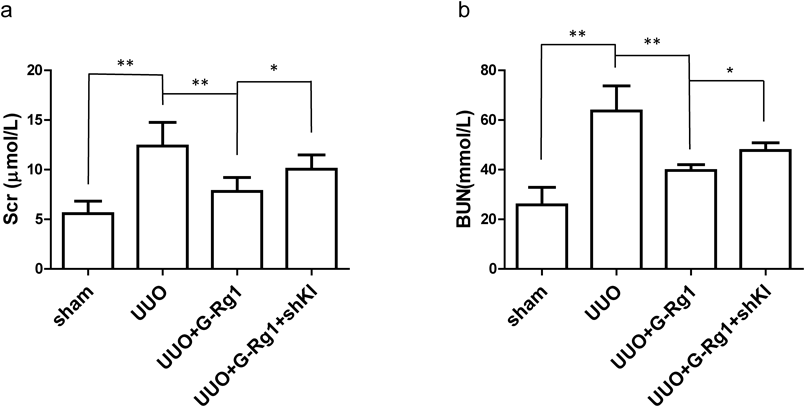
UUO Deteriorates Kidney Function and HistopathologyAfter 14 d, we found that UUO elevated the serum creatinine (Scr) (12.38±2.38 vs. 5.57±1.27, p<0.001) and BUN levels (63.63±10.17 vs. 25.86±7.03, p<0.01), resulting in a deterioration in renal function compared with the control group (Fig. 1). Additionally, renal histological analysis revealed that UUO led to severe tubulointerstitial injures, including interstitial fibrosis, edema, tubular ectasia, necrosis, atrophy and inflammatory cell infiltration (Fig. 2).
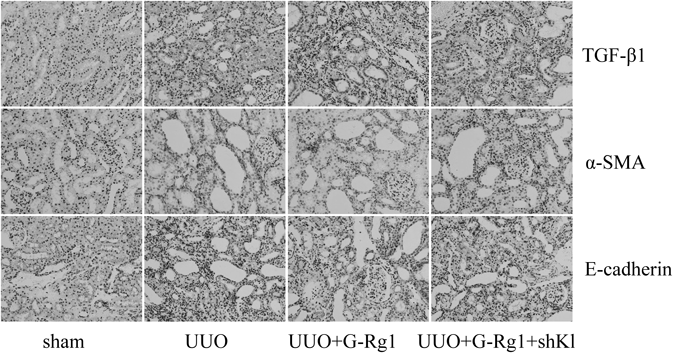
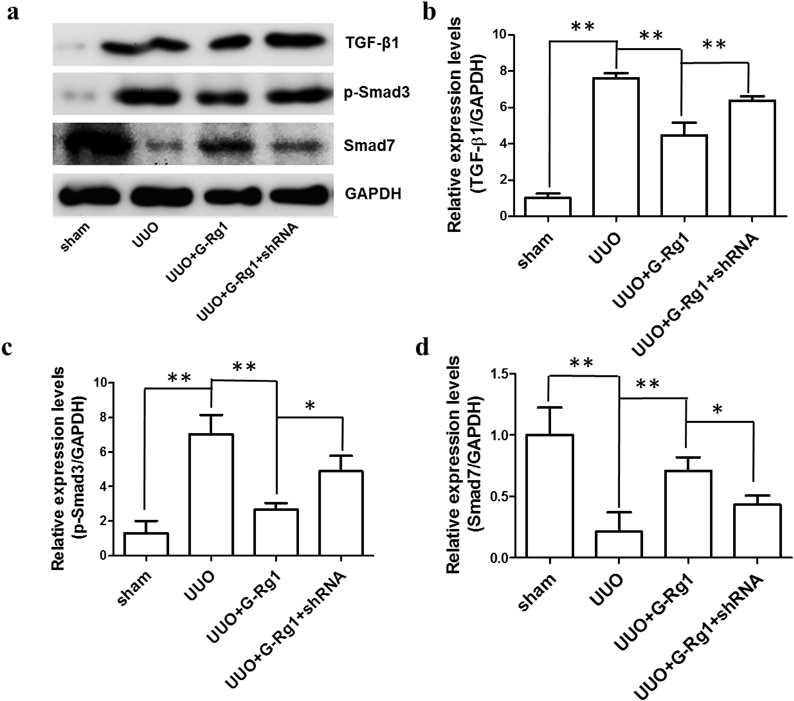
UUO Triggers EMT and Activates TGF-β1/Smad SignalingEMT is an important step in renal fibrosis that can be initiated in fibrotic kidneys, including obstructive nephropathy. We found that UUO caused increased profibrotic TGF-β1 expression, which is a potent EMT inducer. Furthermore, UUO upregulated the mesenchymal marker, α-SMA, and downregulated the epithelial marker, E-cadherin (Fig. 3). TGF-β1 binds to TGF-β1 receptor II, leading to the phosphorylation of Smad proteins, including the receptor-regulated Smad3 and the inhibitory Smad7, and ultimately to the activation of the TGF-β1/Smad signaling pathway. In this study, we observed that UUO significantly increased TGF-β1 and p-Smad3 expression (Figs. 4a–c), while dramatically reducing Smad7 levels (Figs. 4a, d). Together, these data indicate that UUO-induced EMT involves TGF-β1/Smad signaling.
G-Rg1 Improves Kidney Function and Ameliorates TGF-β1-Induced Renal Fibrosis in UUO RatsIn this study, we found that G-Rg1 administration improved kidney function by reducing the level of Scr and BUN (Figs. 1a, b). Moreover, G-Rg1 treatment markedly decreased the UUO-induced TGF-β1 and α-SMA overexpression, and conversely restored E-cadherin levels to a certain extent, as shown by immunohistochemistry (Fig. 3). These data demonstrated that G-Rg1 improved renal function, possibly by inhibiting the UUO-induced EMT responses. To elucidate the mechanism underlying the antifibrotic effect of G-Rg1, we tested the effects of G-Rg1 treatment on TGF-β1, p-Smad3 and Smad7 expression. As shown in Fig. 4, TGF-β1 expression was significantly induced after UUO surgery. In contrast, TGF-β1 expression in the G-Rg1 treatment group was significantly decreased compared with the model group, suggesting that G-Rg1 administration reduced TGF-β1 expression. Similar results were found with p-Smad3 expression, which was also dramatically increased in the model group compared with the sham group, but this UUO-induced increase was significantly attenuated by G-Rg1 administration. Conversely, Smad7 expression was decreased following UUO compared with the control, however G-Rg1 administration reversed the UUO-induced downregulation of Smad7. These results support the hypothesis that G-Rg1 can reverse UUO-induced renal fibrosis, at least partly, through regulating TGF-β1/Smad signaling and the associated EMT process.
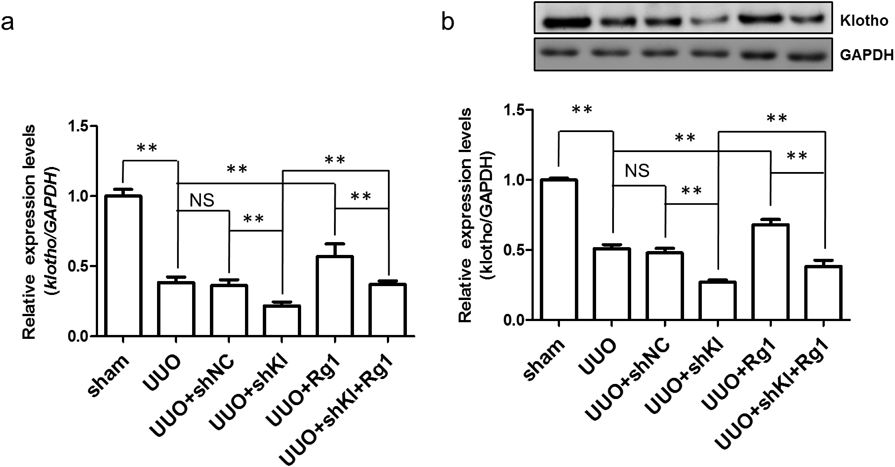
Klotho Is Essential for G-Rg1 in Modulating TGF-β1-induced Renal FibrosisTo clarify the relationship between Klotho and G-Rg1 in alleviating TIF, we knocked down Klotho via Klotho shRNA. Our data showed klotho gene knockdown dramatically reduced the expression of Klotho in the fibrotic kidney both at the mRNA and protein levels, confirming the efficiency of Klotho knockdown (Figs. 5a, b). In addition, Klotho knockdown largely abolished the kidney protective effects of G-Rg1, as the kidney function deteriorated even in the presence of G-Rg1 (Figs. 1, 2). Furthermore, the expression levels of TGF-β1 and α-SMA increased, while the E-cadherin levels decreased (Fig. 3). Consistently, the TGF-β1 downstream signaling showed upregulation of p-Smad3 and downregulation of the inhibitory Smad7 compared with the G-Rg1 treatment group (Fig. 4). This means that Klotho is essential for G-Rg1 in modulating TGF-β1-induced renal fibrosis and the associated EMT process.
G-Rg1 Treatment Upregulates Klotho Expression and Reverses the UUO-Induced Klotho Decrease at the mRNA and Protein LevelsIt has been reported that Klotho downregulation is common in damaged kidneys regardless of etiology.21) We also observed that UUO dramatically downregulated the level of Klotho at the mRNA and protein levels compared with the sham group (Figs. 5a, b). However, it is not clear whether G-Rg1 treatment can upregulate Klotho expression. In this study, we found that G-Rg1 treatment elevated the expression level of Klotho at both the mRNA and protein levels compared with the UUO group (Figs. 5a, b). Thus, Klotho upregulation by G-Rg1 treatment may be used for treatment of kidney injury associated with Klotho deficiency.
DISCUSSION
In the current study, we demonstrated that G-Rg1 ameliorated UUO-induced TIF by a Klotho-dependent mechanism in the kidney, as klotho knockdown abolished most of the beneficial effects. In addition, TGF-β1 and its downstream signaling molecules were suppressed, which was accompanied by changes in EMT markers, due to enhanced Klotho expression in the presence of G-Rg1. Our results suggest that G-Rg1 ameliorates TIF by targeting the Klotho/TGF-β1/Smad pathway, and this protective mode supports the possibility of using G-Rg1 for treating renal fibrosis associated with Klotho deficiency.
The cellular and molecular mechanisms underlying TIF have been extensively studied, however, few antifibrotic therapies are available in clinical practice.22,23) The pathological events associated with TIF are myofibroblast proliferation and activation followed by an accumulation of extracellular matrix.24) Although the specific origin of these myofibroblasts has aroused recent controversy, we believe that it is actually the EMT in renal tubular cells that influences myofibroblasts.25,26) Epithelial cells undergoing EMT lose characteristic epithelial markers, such as E-cadherin and ZO-1, and acquire classic mesenchymal markers, such as α-SMA and FSP-1.27) Previous studies have shown that the level of EMT is closely correlated with the degree of renal fibrogenesis, hence it has been proposed to be an important mechanism of renal fibrosis.28) TGF-β1 is the most pivotal profibrotic driver of EMT induction,29) as has been demonstrated in previous studies by us and others in vitro30,31) and in vivo.32,33) In this signaling pathway, p-Smad3 is markedly activated and functions as the main fibrogenic mediator. Conversely, Smad7, an inhibiting molecule, is decreased during renal fibrosis.32)
In this study, we observed that UUO caused decreased renal function, and led to inflammatory cell infiltration, epithelial vacuolar necrosis, tubular atrophy and TIF. Moreover, we showed by immunohistochemistry that UUO increased TGF-β1 and α-SMA expression, which was accompanied by decreased E-cadherin expression. Alterations in these EMT markers indicated that EMT was triggered by UUO. Furthermore, Western blotting showed that UUO enhanced TGF-β1 and p-Smad3 expression, and suppressed Smad7 expression. These results suggest that the imbalance in the TGF-β/Smad signaling was involved in the progression of renal fibrosis.
Klotho is a novel renoprotective protein that exhibits multiple beneficial functions such as renal fibrogenesis inhibition.34,35) The kidney is the primary organ that expresses Klotho,36) and its deficiency seems to be a biomarker of chronic kidney disease (CKD),37,38) as Klotho deficiency aggravates renal fibrosis and accelerates CKD progression.39) In agreement with this, we have previously shown that renal Klotho expression was decreased by UUO, and interestingly, recombinant Klotho treatment suppressed TIF by inhibiting ER stress.11) Moreover, we have observed an ameliorating effect of recombinant Klotho on TGF-β1-induced EMT in CsA-treated rats.12) Klotho directly bound to TGF-β1 receptor, which was followed by suppression of its downstream fibrotic signaling,10,15) confirming that Klotho deficiency plays an important role in TGF-β1-related renal fibrosis. Targeting Klotho by increasing endogenous Klotho has been observed to ameliorate kidney injury induced by UUO40) or CsA,41) therefore, Klotho has been developed as a therapeutic target in renal fibrosis.
Previous studies have found that G-Rg1 can inhibit cell apoptosis and decrease inflammation,42) as well as attenuate the TGF-β-related EMT process.43) We have also observed that G-Rg1 reduced cell apoptosis and TIF in kidney fibrosis.18,19) G-Rg1 and Klotho are both involved in TIF, and we proposed that the inhibitory effect of G-Rg1 may be related to the Klotho effect. We found that G-Rg1 administration alleviated UUO-associated fibrosis, which was accompanied by alterations in the expression of the TGF-β1/Smad pathway and EMT-related proteins, including upregulation of Smad7, and downregulation of TGF-β1 and p-Smad3. However, klotho knockdown by Klotho shRNA obviated the antifibrotic effects of G-Rg1 and strengthened the TGF-β1/Smad pathway dramatically, indicating that Klotho is essential for the G-Rg1 kidney protective effects and that Klotho downregulation plays a role in the induction of the TGF-β1/Smad pathway. We think that this is a novel mechanism of the protective effects of G-Rg1 in the fibrotic kidney. Another interesting finding in this study was that G-Rg1 application enhanced endogenous Klotho expression both at the mRNA and protein levels. However, to further elucidate the underlying mechanism by which G-Rg1 mediates Klotho expression, further studies are required. Our findings uncovered the beneficial effect of G-Rg1 against the TGF-β1/Smad pathway and the EMT process, and revealed that the effect was Klotho-dependent.
There are several limitations in our study. First, we demonstrated the relationship between G-Rg1 and the Klotho/TGF-β1/Smad pathway, but we did not show direct evidence of EMT, as changes in EMT biomarkers only indirectly indicated EMT. Second, we did not examine the expression of vascular endothelial-cadherin or other markers after G-Rg1 treatment, thus we did not exclude the effects of mesenchymal transition in other cells, such as endothelial cells or resident fibroblasts, on renal fibrosis. Finally, we demonstrated that G-Rg1 elevated Klotho expression, but the exact mechanism remains to be elucidated.
CONCLUSION
In summary, our study demonstrated that UUO activated the TGF-β1-related fibrotic process and decreased Klotho expression, and that G-Rg1 treatment suppressed TGF-β1 signaling, leading to an amelioration of EMT and the fibrotic process via a Klotho-dependent mechanism. Our findings suggest that Klotho-targeted treatments such as G-Rg1 may be developed as an anti-TIF strategy in fibrotic kidney disease related to Klotho deficiency.
Acknowledgments
This study was supported by the Program for the Talents in science and education of Suzhou, Jiangsu Province, China (No. KJXW2014049 and No. KJXW2016061).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Zou XZ, Liu T, Gong ZC, Hu CP, Zhang Z. MicroRNAs-mediated epithelial-mesenchymal transition in fibrotic diseases. Eur. J. Pharmacol., 796, 190–206 (2017).
- 2) Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest., 110, 341–350 (2002).
- 3) Zhao L, Chi L, Zhao J, Wang X, Chen Z, Meng L, Liu G, Guan G, Wang F. Serum response factor provokes epithelial–mesenchymal transition in renal tubular epithelial cells of diabetic nephropathy. Physiol. Genomics, 48, 580–588 (2016).
- 4) Yao J, Ke Z, Wang X, Peng F, Li B, Wu R. Epithelial-mesenchymal transition and apoptosis of renal tubular epithelial cells are associated with disease progression in patients with IgA nephropathy. Molecular Medicine Reports, 10, 39–44 (2014).
- 5) Inoue T, Umezawa A, Takenaka T, Suzuki H, Okada H. The contribution of epithelial–mesenchymal transition to renal fibrosis differs among kidney disease models. Kidney Int., 87, 233–238 (2015).
- 6) Kuro-o M, Matsumura Y, Aizawa H, Kawaguchi H, Suga T, Utsugi T, Ohyama Y, Kurabayashi M, Kaname T, Kume E, Iwasaki H, Iida A, Shiraki-Iida T, Nishikawa S, Nagai R, Nabeshima YI. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature, 390, 45–51 (1997).
- 7) Hu MC, Shi M, Zhang J, Quinones H, Kuro-o M, Moe OW. Klotho deficiency is an early biomarker of renal ischemia–reperfusion injury and its replacement is protective. Kidney Int., 78, 1240–1251 (2010).
- 8) Scholze A, Liu Y, Pedersen L, Xia S, Roth HJ, Hocher B, Rasmussen LM, Tepel M. Soluble alpha-klotho and its relation to kidney function and fibroblast growth factor-23. J. Clin. Endocrinol. Metab., 99, E855–E861 (2014).
- 9) Sugiura H, Yoshida T, Shiohira S, Kohei J, Mitobe M, Kurosu H, Kuro-o M, Nitta K, Tsuchiya K. Reduced Klotho expression level in kidney aggravates renal interstitial fibrosis. Am. J. Physiol. Renal Physiol., 302, F1252–F1264 (2012).
- 10) Zhou L, Li Y, Zhou D, Tan RJ, Liu Y. Loss of Klotho contributes to kidney injury by derepression of Wnt/beta-catenin signaling. J. Am. Soc. Nephrol., 24, 771–785 (2013).
- 11) Liu QF, Ye JM, Deng ZY, Yu LX, Sun Q, Li SS. Ameliorating effect of Klotho on endoplasmic reticulum stress and renal fibrosis induced by unilateral ureteral obstruction. Iran. J. Kidney Dis., 9, 291–297 (2015).
- 12) Liu QF, Ye JM, Yu LX, Dong XH, Feng JH, Xiong Y, Gu XX, Li SS. Klotho mitigates cyclosporine A (CsA)-induced epithelial–mesenchymal transition (EMT) and renal fibrosis in rats. Int. Urol. Nephrol., 49, 345–352 (2017).
- 13) Doi S, Masaki T. Klotho as a therapeutic target during the development of renal fibrosis. Contrib. Nephrol., 189, 178–183 (2017).
- 14) Irifuku T, Doi S, Sasaki K, Doi T, Nakashima A, Ueno T, Yamada K, Arihiro K, Kohno N, Masaki T. Inhibition of H3K9 histone methyltransferase G9a attenuates renal fibrosis and retains klotho expression. Kidney Int., 89, 147–157 (2016).
- 15) Doi S, Zou Y, Togao O, Pastor JV, John GB, Wang L, Shiizaki K, Gotschall R, Schiavi S, Yorioka N, Takahashi M, Boothman DA, Kuro-o M. Klotho inhibits transforming growth factor-beta1 (TGF-beta1) signaling and suppresses renal fibrosis and cancer metastasis in mice. J. Biol. Chem., 286, 8655–8665 (2011).
- 16) Cheng Y, Shen LH, Zhang JT. Anti-amnestic and anti-aging effects of ginsenoside Rg1 and Rb1 and its mechanism of action. Acta Pharmacol. Sin., 26, 143–149 (2005).
- 17) Lee CH, Kim JH. A review on the medicinal potentials of ginseng and ginsenosides on cardiovascular diseases. Journal of Ginseng Research, 38, 161–166 (2014).
- 18) Liu QF, Deng ZY, Ye JM, He AL, Li SS. Ginsenoside Rg1 protects chronic cyclosporin a nephropathy from tubular cell apoptosis by inhibiting endoplasmic reticulum stress in rats. Transplant. Proc., 47, 566–569 (2015).
- 19) Li SS, Ye JM, Deng ZY, Yu LX, Gu XX, Liu QF. Ginsenoside-Rg1 inhibits endoplasmic reticulum stress-induced apoptosis after unilateral ureteral obstruction in rats. Ren. Fail., 37, 890–895 (2015).
- 20) Chen LJ, Cheng MF, Ku PM, Cheng JT. Cerebral klotho protein as a humoral factor for maintenance of baroreflex. Horm. Metab. Res., 47, 125–132 (2015).
- 21) Hu MC, Kuro-o M, Moe OW. The emerging role of Klotho in clinical nephrology. Nephrol. Dial. Transplant., 27, 2650–2657 (2012).
- 22) Nogueira A, Pires MJ, Oliveira PA. Pathophysiological mechanisms of renal fibrosis: a review of animal models and therapeutic strategies. In Vivo, 31, 1–22 (2017).
- 23) Boor P, Sebekova K, Ostendorf T, Floege J. Treatment targets in renal fibrosis. Nephrol. Dial. Transplant., 22, 3391–3407 (2007).
- 24) Hills CE, Squires PE. TGF-beta1-induced epithelial-to-mesenchymal transition and therapeutic intervention in diabetic nephropathy. Am. J. Nephrol., 31, 68–74 (2010).
- 25) Zeisberg M, Duffield JS. Resolved: EMT produces fibroblasts in the kidney. J. Am. Soc. Nephrol., 21, 1247–1253 (2010).
- 26) LeBleu VS, Taduri G, O’Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med., 19, 1047–1053 (2013).
- 27) Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol., 15, 178–196 (2014).
- 28) Schelling JR. Tubular atrophy in the pathogenesis of chronic kidney disease progression. Pediatr. Nephrol., 31, 693–706 (2016).
- 29) Meng XM, Tang PM, Li J, Lan HY. TGF-beta/Smad signaling in renal fibrosis. Frontiers in Physiology, 6, 82 (2015).
- 30) Li SS, Liu QF, He AL, Wu FR. Tranilast attenuates TGF-beta1-induced epithelial–mesenchymal transition in the NRK-52E cells. Pak. J. Pharm. Sci., 27, 51–55 (2014).
- 31) Qi F, Cai P, Liu X, Peng M, Si G. Adenovirus-mediated P311 inhibits TGF-beta1-induced epithelial–mesenchymal transition in NRK-52E cells via TGF-beta1-Smad-ILK pathway. Bioscience Trends, 9, 299–306 (2015).
- 32) Jia L, Ma X, Gui B, Ge H, Wang L, Ou Y, Tian L, Chen Z, Duan Z, Han J, Fu R. Sorafenib ameliorates renal fibrosis through inhibition of TGF-beta-induced epithelial-mesenchymal transition. PLOS ONE, 10, e0117757 (2015).
- 33) Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Transforming growth factor-beta regulates tubular epithelial-myofibroblast transdifferentiation in vitro. Kidney Int., 56, 1455–1467 (1999).
- 34) Kuro-o M. The FGF23 and Klotho system beyond mineral metabolism. Clin. Exp. Nephrol., 21 (Suppl. 1), 64–69 (2017).
- 35) Izquierdo MC, Perez-Gomez MV, Sanchez-Nino MD, Sanz AB, Ruiz-Andres O, Poveda J, Moreno JA, Egido J, Ortiz A. Klotho, phosphate and inflammation/ageing in chronic kidney disease. Nephrol. Dial. Transplant., 27 (Suppl. 4), iv6–iv10 (2012).
- 36) Hu MC, Shi M, Zhang J, Addo T, Cho HJ, Barker SL, Ravikumar P, Gillings N, Bian A, Sidhu SS, Kuro-o M, Moe OW. Renal production, uptake, and handling of circulating alphaKlotho. J. Am. Soc. Nephrol., 27, 79–90 (2016).
- 37) Park MY, Herrmann SM, Saad A, Eirin A, Tang H, Lerman A, Textor SC, Lerman LO. Biomarkers of kidney injury and klotho in patients with atherosclerotic renovascular disease. Clin. J. Am. Soc. Nephrol., 10, 443–451 (2015).
- 38) Pavik I, Jaeger P, Ebner L, Wagner CA, Petzold K, Spichtig D, Poster D, Wuthrich RP, Russmann S, Serra AL. Secreted Klotho and FGF23 in chronic kidney disease Stage 1 to 5: a sequence suggested from a cross-sectional study. Nephrol. Dial. Transplant., 28, 352–359 (2013).
- 39) Kim HR, Nam BY, Kim DW, Kang MW, Han JH, Lee MJ, Shin DH, Doh FM, Koo HM, Ko KI, Kim CH, Oh HJ, Yoo TH, Kang SW, Han DS, Han SH. Circulating alpha-klotho levels in CKD and relationship to progression. Am. J. Kidney Dis., 61, 899–909 (2013).
- 40) Zhang Q, Liu L, Lin W, Yin S, Duan A, Liu Z, Cao W. Rhein reverses Klotho repression via promoter demethylation and protects against kidney and bone injuries in mice with chronic kidney disease. Kidney Int., 91, 144–156 (2017).
- 41) Hu Y, Mou L, Yang F, Tu H, Lin W. Curcumin attenuates cyclosporine A induced renal fibrosis by inhibiting hypermethylation of the klotho promoter. Molecular Medicine Reports, 14, 3229–3236 (2016).
- 42) Gao Y, Chu S, Zhang Z, Chen N. Hepataprotective effects of ginsenoside Rg1—A review. J. Ethnopharmacol., 206, 178–183 (2017).
- 43) Xie XS, Yang M, Liu HC, Zuo C, Li HJ, Fan JM. Ginsenoside Rg1, a major active component isolated from Panax notoginseng, restrains tubular epithelial to myofibroblast transition in vitro. J. Ethnopharmacol., 122, 35–41 (2009).