Abstract
Our previous studies have shown that phenylephrine-induced contraction of cutaneous arteries is primarily mediated via α1A-adrenoceptors, but not α1D-adrenoceptors that generally mediate vascular contraction, and that the larger part of the contraction is induced in a voltage-dependent Ca2+ channel (VDCC)-independent manner. Here, we investigated the mechanism underlying the smaller part of the α1A-adrenoceptor-mediated contraction, i.e., VDCC-dependent one, in cutaneous arteries. Isometric contraction was measured with wire myograph in endothelium-denuded tail and iliac arterial rings isolated from male Wistar rats. LOE908 (10 µM), a cation channel blocker, partially inhibited the contraction induced by phenylephrine in tail and iliac arteries. Nifedipine (10 µM) further suppressed the phenylephrine-induced contraction that remained in the presence of LOE908 (10 µM) in iliac arteries but barely in tail arteries, suggesting that phenylephrine-induced depolarization in tail arteries is due to the activation of LOE908-sensitive cation channels. In iliac arteries, the contraction induced by A-61603, a specific α1A-adrenoceptor agonist, was also partially inhibited by LOE908 (10 µM); however, nifedipine had little effect on the A-61603-induced contraction that remained in the presence of LOE908 (10 µM), suggesting that depolarization mediated via α1A-adrenoceptors is due to the activation of LOE908-sensitive cation channels even in iliac arteries. These results suggest that membrane depolarization mediated via α1Α-adrenoceptors is caused by cation influx through LOE908-sensitive cation channels. Less contribution of VDCC to phenylephrine-induced contraction in tail arteries compared to in iliac arteries is likely due to that α1Α-adrenoceptor-mediated activation of VDCC is caused only by depolarization via cation influx through LOE908-sensitive cation channels.
INTRODUCTION
Cutaneous arteries have a unique property to respond to changes in temperature: Low temperature causes the enhancement of contraction.1) Although α2C-adrenoceptors have been proposed to be involved in the enhanced contractile response,2–4) our previous studies showed that α1A-adrenoceptors also contribute to the enhancement at low temperature in rat tail arteries.5) In addition, a well correlation between the enhanced contractile response to phenylephrine at low temperature and the expression level of α1A-adrenoceptors was accompanied with a less contribution of voltage-dependent Ca2+ channels (VDCC) to the contraction induced by phenylephrine in rat tail arteries.6) VDCC is well known to be suppressed at low temperature.7,8) Thus, the enhancement of phenylephrine-induced contraction at low temperature in cutaneous arteries is likely due to a less contribution of VDCC to the contraction mediated via α1A-adrenoceptors. These findings suggest an essential role of α1A-adrenoceptors in the function of cutaneous arteries. On the other hand, we also found that VDCC contributes to the part of contraction mediated via α1A-adrenoceptors in rat tail arteries.5)
The tail artery plays a role for thermoregulation as a typical cutaneous artery, whereas the iliac artery is a visceral artery and supplies blood to the pelvis, legs and tail. The expression patterns of α1-adrenoceptor subtypes have been shown to be different between these arteries of Wistar rats; α1A-adrenoceptors are prominent in the tail artery, whereas both α1A- and α1D-subtypes are expressed at similar levels in the iliac artery.9) In the present study, we tried to elucidate the mechanism of membrane depolarization underlying contraction mediated via α1A-adrenoceptors in isolated rat tail and iliac arteries.
MATERIALS AND METHODS
Measurement of ContractionMale Wistar rats (8–12 weeks old; SLC, Shizuoka, Japan) were housed in a 12 h light–dark cycle, with food and water available ad libitum, and treated as approved by the Institutional Animal Care and Use Committee (approval number 136055) and according to the Guidelines for Animal Experiments established by the Japanese Pharmacological Society. The rats were treated with sodium pentobarbital (50 mg/kg, intraperitoneally) and killed by decapitation. Common iliac artery and tail artery were isolated in ice-cold Krebs–Henseleit (KH) solution (in mM: NaCl, 118; KCl, 4.7; CaCl2, 2.55; MgSO4, 1.18; KH2PO4, 1.18; NaHCO3, 24.8; and glucose, 11.1) aerated with 95% O2/5% CO2. The isolated tail artery was kept in KH solution overnight at 4°C to prevent spontaneous contractions. The isolated arteries were cut into ring segments around 2 mm in width. The outer diameter of the tail and iliac arteries was around 0.6 and 1.0–1.2 mm, respectively. Endothelial cells in each segment was removed by scraping the lumen with silk ligature. Each ring segment was mounted on a myograph (Multi Myograph Model 610M; Danish Myo Technology A/S, Aarhus, Denmark) with two tungsten wires of 40 µm in diameter in KH solution aerated with 95% O2/5% CO2 at 37°C. Isometric tension was measured and recorded using a data analysis program (Myodaq 2.01; Danish Myo Technology A/S). Tail and iliac arteries were loaded with a resting tension of 5–10 mN. After a suitable stabilization period, KH solution was replaced to an 80-mM KCl solution (in mM: NaCl, 42.7; KCl, 80; CaCl2, 2.55; MgSO4, 1.18; KH2PO4, 1.18; NaHCO3, 24.8; and glucose, 11.1). This procedure was repeated until stable contraction was attained. We used only specimens in which acetylcholine (1 µM) induced a relaxation of less than 10% on the pre-contraction induced by noradrenaline (100 nM). Phenylephrine or A-61603 was applied in a cumulative manner. Each contraction is normalized by 80-mM KCl-induced contraction. The concentration–response curve was fitted by Hill’s equation using Origin software (OriginLab, Northampton, MA, U.S.A.).
Statistical AnalysisData are expressed as mean ± standard error of the mean (S.E.M.) and were statistically analyzed by paired or unpaired t-test. p Value less than 0.05 was considered significant.
DrugsAcetylcholine chloride, phenylephrine hydrochloride, nifedipine and noradrenaline were purchased from Sigma-Aldrich (St. Louis, MO, U.S.A.), 3,4-dihydro-6,7-dimethoxy-α-phenyl-N,N-bis[2-(2,3,4-trimethoxyphenyl)-ethyl]-1-isoquinolineacetamide hydrochloride (LOE908) and A-61603 (Cayman chemical, Ann Arbor, MI, U.S.A.) was purchased from Funakoshi (Osaka, Japan) and other chemicals of special grade were from FUJIFILM Wako Pure Chemical (Osaka, Japan). Acetylcholine chloride, phenylephrine hydrochloride, and noradrenaline were dissolved in distilled water and nifedipine, LOE908, and A-61603 were dissolved in dimethyl sulfoxide.
RESULTS
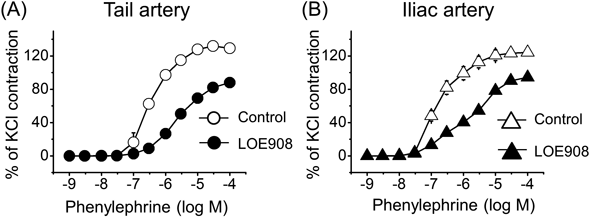
Our previous study showed that the contribution of VDCC to phenylephrine induced-contraction in tail arteries is much less than in iliac arteries.6) Membrane depolarization that activates VDCC would be induced by the activation of cation channels. We thus investigated the effect of LOE908, a cation channel inhibitor, on phenylephrine-induced contraction. LOE908 at 10 µM, which has been shown to inhibit several types of non-selective cation channels,10) shifted the concentration–response curve of phenylephrine to the right and attenuated the maximum contraction in both tail and iliac arteries (Figs. 1A, B, Table 1). Nifedipine (10 µM), a VDCC inhibitor, barely affected the concentration–response curve of phenylephrine in the presence of LOE908 (10 µM) in tail arteries (Fig. 2A, Table 2). In contrast, nifedipine (10 µM) shifted the concentration–response curve of phenylephrine in the presence of LOE908 (10 µM) to the right and attenuated the maximum contraction in iliac arteries (Fig. 2B, Table 2). Although the concentration of nifedipine used in the present study, i.e., 10 µM, might be relatively high, our preliminary experiments showed that nifedipine at 1 µM only partially suppressed phenylephrine-induced contraction and at 10 µM largely suppressed it in rat iliac arteries. Moreover, we previously showed that nifedipine even at 10 µM only partially inhibits phenylephrine-induced contraction in rat tail arteries.6) Thus, this concentration of nifedipine is likely suitable for fully inhibiting the contraction mediated by VDCC activation.
Table 1. Parameters for Concentration–Response Curves of Phenylephrine in the Absence and Presence of LOE908 (10 µM) in Tail and Iliac Arteries
| Tail artery (n = 5) | Iliac artery (n = 4) |
|---|
| Control | LOE908 | Control | LOE908 |
|---|
| Max | 128.1 ± 3.9 | 89.5 ± 4.9b) | 120.9 ± 4.9 | 98.0 ± 5.02a) |
| pEC50 | 6.46 ± 0.08 | 5.60 ± 0.45b) | 6.75 ± 0.06 | 5.82 ± 0.11b) |
The maximum contraction (Max) was normalized to the contraction induced by 80 mM KCl (23.7 ± 1.9 and 21.3 ± 2.9 mN in tail and iliac arteries, respectively). pEC50 was negative log of the half-maximally effective concentration. Data are expressed as mean ± S.E.M. and were statistically analyzed by paired t-test. a) p < 0.01, b) p < 0.05 vs. corresponding control.
Table 2. Parameters for Concentration–Response Curves of Phenylephrine in the Presence of LOE908 and Presence of Combination of LOE908 (10 µM) and Nifedipine (10 µM) in Tail and Iliac Arteries
| Tail artery | Iliac artery |
|---|
| LOE908 (n = 4) | LOE908/nifedipine (n = 5) | LOE908 (n = 5) | LOE908/nifedipine (n = 5) |
|---|
| Max | 85.3 ± 5.9 | 95.9 ± 4.6 | 106.0 ± 6.2 | 59.6 ± 5.1b) |
| pEC50 | 5.61 ± 0.05 | 5.63 ± 0.05 | 6.02 ± 0.28 | 5.05 ± 0.12a) |
The maximum contraction (Max) was normalized to the contraction induced by 80 mM KCl (25.4 ± 2.0 and 23.8 ± 1.3 mN in tail and iliac arteries, respectively). pEC50 was negative log of the half-maximally effective concentration. Data are expressed as mean ± S.E.M. and were statistically analyzed by unpaired t-test. a) p < 0.01, b) p < 0.05 vs. corresponding control.
The effect of LOE908 (10 µM) on the contraction induced by A-61603, an α1A-adrenoceptor selective agonist,11) was further investigated in iliac arteries. LOE908 (10 µM) shifted the concentration–response curve of A-61603 to the right with a significant increase in EC50 and significantly attenuated the maximum contraction in iliac arteries (Fig. 3A, Table 3). Nifedipine (10 µM) barely affected the concentration–response curve of A-61603 in the presence of LOE908 (10 µM; Fig. 3B, Table 3).
Table 3. Parameters for Concentration–Response Curves of A-61603 in the Absence and Presence of LOE908 (10 µM) and Combination of LOE908 (10 µΜ) and Nifedipine (10 µM) in Iliac Arteries
| Figure 3A (n = 4) | Figure 3B |
|---|
| Control | LOE908 | LOE908 (n = 6) | LOE908 and nifedipine (n = 7) |
|---|
| Max | 122.9 ± 8.7 | 98.2 ± 5.6b) | 102.3 ± 4.3 | 103.7 ± 4.2 |
| pEC50 | 7.96 ± 0.10 | 7.46 ± 0.10a) | 7.60 ± 0.09 | 7.38 ± 0.04 |
The maximum contraction (Max) was normalized to the contraction induced by 80 mM KCl (22.1 ± 1.9 mN). pEC50 was negative log of the half-maximally effective concentration. Data are expressed as mean ± S.E.M. and were statistically analyzed by paired (Fig. 3A) or unpaired t-test (Fig. 3B). a) p < 0.01, b) p < 0.05 vs. corresponding control.
DISCUSSION
The present study investigated the mechanism of α1A-adrenoceptor-mediated, VDCC-dependent vascular contraction. The results obtained here suggest that the depolarization induced by the activation of α1A-adrenoceptors is due to cation influx through LOE908-sensitive cation channels in both rat tail and iliac arteries.
LOE908, which inhibits several types of non-selective cation channels vide infra, partly suppressed phenylephrine-induced contraction in rat iliac and tail arteries. Interestingly, there was apparent difference in the remaining LOE908-insensitive contraction between the arteries; nifedipine barely affected the contraction in tail arteries, but further suppressed it in iliac arteries. We have previously shown that LOE908 at 10 µM had no effect on intracellular Ca2+ elevation induced by U46619, a prostanoid TP receptor agonist, in the presence of verapamil, a voltage-dependent Ca2+ channel inhibitor, in rat aorta.10) LOE908-sensitive cation channels activated via TP receptors and α1-adrenoceptors were suggested to be less Ca2+ permeable and evoke membrane depolarization in rat tail arteries and aorta. LOE908 has also been shown to inhibit vasopressin-activated, non-selective cation channels in A7r5 cells, a smooth muscle cell line derived from rat aorta,12) and to inhibit nifedipine-insensitive contraction and intracellular Ca2+ elevation induced by endothelin-1 and vasopressin in rat aorta.13) Endothelin, TP, adrenaline α1, and vasopressin V1A receptors are all Gq-coupled receptors, producing inositol 1,4,5-trisphosphatase and diacylglycerol (DAG) by activating phosphatidylinositol-specific phospholipase Cβ. However, the effects of LOE908 on the intracellular Ca2+ elevation and contraction induced by Gq-coupled receptor agonists were various. Thus, the interaction between α1-adrenoceptors and LOE908-sensitive cation channels might be different between rat iliac and tail arteries.
Our previous studies have suggested that the contribution of VDCC to α1A-adrenoceptor-meidated contraction is less than that to α1D-adrenoceptor-mediated one and that phenylephrine-induced contraction is mediated via mostly α1A-adrenoceptors in tail arteries and via both α1A- and α1D-adrenoceptors in iliac arteries.5,6) Martí et al. have demonstrated using quantitative RT-PCR that both α1A- and α1D-adrenoceptors are expressed in a similar level in iliac arteries, whereas α1A-adrenoceptors are highly expressed in tail arteries.9) The present study showed that nifedipine barely affected the contraction induced by the α1A-adrenoceptor selective agonist A-61603 in the presence of LOE908 in iliac arteries. In contrast, nifedipine further suppressed the LOE908-insensitive contraction induced by phenylephrine, which stimulates both α1A- and α1D-adrenoceptors, in the same arteries. It is thus plausible to say that the membrane depolarization induced by α1A-adrenoceptor stimulation is mediated via LOE908-sensitive cation channels, which are barely permeable to Ca2+, whereas that induced by α1D-adrenoceptor stimulation is mediated through an LOE908-insensitive mechanism. Martí et al. also showed that α1A-adrenoceptors are predominated in rat small mesenteric arteries, whereas α1D-adrenoceptors are predominated in rat aorta.9) Thus, LOE908-sensitive cation channels may also be involved in the contraction in rat small mesenteric arteries, but not in rat aorta.
Several studies have suggested that LOE908 inhibits store-operated Ca2+ channels, transient receptor potential canonical 1 (TRPC1), transient receptor potential polycystin 1 (TRPP1), and transient receptor potential melastin 7 (TRPM7).13–16) LOE908 has also been shown to suppress cation channels sensitive to SK&F96365, a receptor-operated channel inhibitor, in HL-60 cells.17) These LOE908-sensitive channels are known to induce Ca2+ influx enough to increase intracellular Ca2+ level. These cation channels are thus unlikely to be LOE908-sensitive non-selective cation channels which are barely permeable to Ca2+ and activated by α1A-adrenoceptor stimulation.
TRPM4 and TRPC6 activated by mechanical stretch have been shown to be involved in membrane depolarization that induces myogenic tone in cerebral arteries.18,19) TRPC1 and TRPC6 activated by α1-adrenoceptor stimulation have also been shown to evoke membrane depolarization, which led to the activation of VDCC.18,20) The involvement of TRPC6 in the contraction of rat aorta induced by Gq-coupled receptor agonists has been suggested: cilostazole, a phosphodiesterase 3 inhibitor, suppressed Ca2+ influx into aortic smooth muscle cells, which was accompanied with increased phosphorylation of TRPC6, and also suppressed angiotensin II-induced contraction of reconstituted ring with rat aortic smooth muscle cells, which was abolished by the expression of a phosphorylation-deficient mutant of TRPC6. Interestingly, angiotensin II-induced contraction of rat aorta was also abolished by nitrendipine, a VDCC inhibitor.21) Thus, angiotensin II-induced contraction of rat aorta is likely to be mediated by VDCC, possibly activated by the membrane depolarization due to TRPC6 activation. Although TRPC6 is also known to induce Ca2+ influx, the Ca2+ influx might not be enough to evoke contraction. These properties are similar to those of LOE908-sensitive contraction shown in the present study. TRPC6 has been shown to be activated by α1A-adrenoceptor stimulation in the PC12 pheochromocytoma cell line.22) Although the functional expression of TRPC6 in rat iliac and tail arteries remains to be confirmed, it has been shown in rat other arteries, i.e., caudal and renal arteries.23,24) Thus, TRPC6 might be a candidate of LOE908-sensitive non-selective cation channels which are barely permeable to Ca2+ and activated by α1A-adrenoceptor stimulation. Further experiments are required to identify the cation channels involved in membrane depolarization evoked by α1A-adrenoceptor stimulation.
In summary, the present study suggests that α1Α-adrenoceptor-mediated activation of VDCC is caused only by membrane depolarization due to cation influx through LOE908-sensitive cation channels. This is the first report, to our knowledge, to provide evidence for the mechanism for α1A-adrenoceptor-mediated depolarization in tail and iliac arterial smooth muscle cells. Less contribution of VDCC to phenylephrine-induced contraction in tail arteries compared to in iliac arteries, shown in our previous study,5,6) might be due to that phenylephrine-induced activation of VDCC is caused only by depolarization via cation influx through the activation of LOE908-sensitive non-selective cation channels via α1A-adrenoceptors in tail arteries.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Cooke JP, Marshall JM. Mechanisms of Raynaud’s disease. Vasc. Med., 10, 293–307 (2005).
- 2) Bailey SR, Mitra S, Flavahan S, Flavahan NA. Reactive oxygen species from smooth muscle mitochondria initiate cold-induced constriction of cutaneous arteries. Am. J. Physiol. Heart Circ. Physiol., 289, H243–H250 (2005).
- 3) Bailey SR, Eid AH, Mitra S, Flavahan S, Flavahan NA. Rho kinase mediates cold-induced constriction of cutaneous arteries: role of a2C-adrenoceptor translocation. Circ. Res., 94, 1367–1374 (2004).
- 4) Chotani MA, Flavahan S, Mitra S, Daunt D, Flavahan NA. Silent α2C-adrenergic receptors enable cold-induced vasoconstriction in cutaneous arteries. Am. J. Physiol. Heart Circ. Physiol., 278, H1075–H1083 (2000).
- 5) Ishida H, Saito S, Ishikawa T. α1A-Adrenoceptors, but not α1B- or α1D-adrenoceptors, contribute to enhanced contractile response to phenylephrine in cooling conditions in the rat tail artery. Eur. J. Pharmacol., 838, 120–128 (2018).
- 6) Ishida H, Saito S, Hishinuma E, Kitayama T, Ishikawa T. Differential contribution of calcium channels to α1-adrenoceptor-mediated contraction is responsible for diverse responses to cooling between rat tail and iliac arteries. Eur. J. Pharmacol., 826, 9–16 (2018).
- 7) Klöckner U, Schiefer A, Isenberg G. L-type Ca-channels: similar Q10 of Ca-, Ba- and Na-conductance points to the importance of ion-channel interaction. Pflugers Arch., 415, 638–641 (1990).
- 8) Peloquin JB, Doering CJ, Rehak R, McRory JE. Temperature dependence of Cav1.4 calcium channel gating. Neuroscience, 151, 1066–1083 (2008).
- 9) Martí D, Miquel R, Ziani K, Gisbert R, Ivorra MD, Anselmi E, Moreno L, Villagrasa V, Barettino D, D’Ocon P. Correlation between mRNA levels and functional role of α1-adrenoceptor subtypes in arteries: evidence of α1L as a functional isoform of the α1A-adrenoceptor. Am. J. Physiol. Heart Circ. Physiol., 289, H1923–H1932 (2005).
- 10) Suzuki K, Saito S, Ishikawa T. Involvement of phosphatidylcholine-specific phospholipase C in thromboxane A2 receptor-mediated extracellular Ca2+ influx in rat aorta. Eur. J. Pharmacol., 677, 123–130 (2012).
- 11) Knepper SM, Buckner SA, Brune ME, DeBernardis JF, Meyer MD, Hancock AA. A-61603, a potent α1-adrenergic receptor agonist, selective for the alpha 1A receptor subtype. J. Pharmacol. Exp. Ther., 274, 97–103 (1995).
- 12) Krautwurst D, Degtiar VE, Schultz G, Hescheler J. The isoquinoline derivative LOE 908 selectively blocks vasopressin-activated nonselective cation currents in A7r5 aortic smooth muscle cells. Naunyn Schmiedebergs Arch. Pharmacol., 349, 301–307 (1994).
- 13) Furutani H, Zhang X-F, Iwamuro Y, Lee K, Okamoto Y, Takikawa O, Fukao M, Masaki T, Miwa S. Ca2+ entry channels involved in contractions of rat aorta induced by endothelin-1, noradrenaline, and vasopressin. J. Cardiovasc. Pharmacol., 40, 265–276 (2002).
- 14) Encabo A, Romanin C, Birke FW, Kukovetz WR, Groschner K. Inhibition of a store-operated Ca2+ entry pathway in human endothelial cells by the isoquinoline derivative LOE 908. Br. J. Pharmacol., 119, 702–706 (1996).
- 15) Hanano T, Hara Y, Shi J, Morita H, Umebayashi C, Mori E, Sumimoto H, Ito Y, Mori Y, Inoue R. Involvement of TRPM7 in cell growth as a spontaneously activated Ca2+ entry pathway in human retinoblastoma cells. J. Pharmacol. Sci., 95, 403–419 (2004).
- 16) Berrout J, Jin M, O’Neil RG. Critical role of TRPP2 and TRPC1 channels in stretch-induced injury of blood–brain barrier endothelial cells. Brain Res., 1436, 1–12 (2012).
- 17) Krautwurst D, Hescheler J, Arndts D, Lösel W, Hammer R, Schultz G. Novel potent inhibitor of receptor-activated nonselective cation currents in HL-60 cells. Mol. Pharmacol., 43, 655–659 (1993).
- 18) Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ. Res., 95, 922–929 (2004).
- 19) Kim E-C, Choi S-K, Lim M, Yeon S-I, Lee Y-H. Role of endogenous ENaC and TRP channels in the myogenic response of rat posterior cerebral arteries. PLOS ONE, 8, e84194 (2013).
- 20) Brayden JE, Earley S, Nelson MT, Reading S. Transient receptor potential (TRP) channels, vascular tone and autoregulation of cerebral blood flow. Clin. Exp. Pharmacol. Physiol., 35, 1116–1120 (2008).
- 21) Nishioka K, Nishida M, Ariyoshi M, Jian Z, Saiki S, Hirano M, Nakaya M, Sato Y, Kita S, Iwamoto T, Hirano K, Inoue R, Kurose H. Cilostazol suppresses angiotensin II-induced vasoconstriction via protein kinase A-mediated phosphorylation of the transient receptor potential canonical 6 channel. Arterioscler. Thromb. Vasc. Biol., 31, 2278–2286 (2011).
- 22) Suzuki F, Morishima S, Tanaka T, Muramatsu I. Snapin, a new regulator of receptor signaling, augments α1A-adrenoceptor-operated calcium influx through TRPC6. J. Biol. Chem., 282, 29563–29573 (2007).
- 23) Salomonsson M, Braunstein TH, Holstein-Rathlou N-H, Jensen LJ. Na+-independent, nifedipine-resistant rat afferent arteriolar Ca2+ responses to noradrenaline: possible role of TRPC channels. Acta Physiol. (Oxf.), 200, 265–278 (2010).
- 24) Mita M, Ito K, Taira K, Nakagawa J, Walsh MP, Shoji M. Attenuation of store-operated Ca2+ entry and enhanced expression of TRPC channels in caudal artery smooth muscle from Type 2 diabetic Goto–Kakizaki rats. Clin. Exp. Pharmacol. Physiol., 37, 670–678 (2010).