Abstract
RNase He1 is a guanylic acid-specific ribonuclease of the RNase T1 family from Hericium erinaceus (Japanese name: Yamabushitake). Its RNA degrading activity is strongly inhibited by Zn2+, similar to other T1 family RNases. However, RNase He1 shows little inhibition of human tumor cell proliferation, unlike RNase Po1, another T1 family RNase from Pleurotus ostreatus (Japanese name: Hiratake). Here, we determined the three-dimensional X-ray crystal structure of RNase He1 in complex with Zn, which revealed that Zn binding most likely prevents substrate entry into the active site due to steric hindrance. This could explain why RNase He1 and other T1 family RNases are inhibited by Zn. The X-ray crystal structures revealed that RNase He1 and RNase Po1 are almost identical in their catalytic sites and in the cysteine residues involved in disulfide bonds that increase their stability. However, our comparison of the electrostatic potentials of their molecular surfaces revealed that RNase He1 is negative whereas RNase Po1 is positive; thus, RNase He1 may not be able to electrostatically bind to the plasma membrane, potentially explaining why it does not exhibit antitumor activity. Hence, we suggest that the cationic characteristics of RNase Po1 are critical to the anti-tumor properties of the protein.
INTRODUCTION
The T1 ribonuclease (RNase) family members are reportedly only present in fungi and bacteria, with RNase T1 from Aspergillus oryzae being the best-known member.1,2) The T1 RNase family members hydrolyze single-stranded RNA via a 2′,3′-cyclic phosphate intermediate at the 3′-terminus of oligonucleotides, and the resultant products are oligonucleotides or mononucleotides with a terminal 3′-phosphate.
We recently characterized two T1 RNases family members from edible mushrooms. One of them was RNase Po1 from Pleurotus ostreatus (Japanese name: Hiratake). RNase Po1 has 101 amino acid residues with a molecular weight of 10789 Da.3) We showed that RNase Po1 is capable of inhibiting human tumor cell line proliferation. RNase Po1 is the first RNase from among the typical members of the RNase T1 family that exhibits antitumor activity.4) The other T1 RNase family member was RNase He1 from Hericium erinaceus (Japanese name: Yamabushitake). RNase He1 contains 100 amino acid residues with a molecular weight of 10690 Da. A multiple sequence alignment of RNase He1 revealed a higher sequence identity with RNase Po1 (60%) than RNase T1 (40%), with the enzyme’s active site well conserved. RNase He1 and RNase Po1 contain six conserved cysteine residues (two more than other T1 family RNases from fungi). RNase He1 was more thermostable than RNase T1, similar to RNase Po1 (Fig. 1). However, RNase He1 does not inhibit human tumor cell line proliferation similar to that exhibited by RNase T1 and other T1 family members.5) A comparison of the enzymatic properties of RNase He1 and RNase Po1 revealed that the optimum pH for enzyme activity of RNase Po1 is 7.5, which is the optimum for most T1 family RNases; however, the optimum pH of RNase He1 is 4.5. The pI of RNase He1 is 4.2, which is similar to that of RNase T1 (pI: 2.9) and most T1 family RNases, while the pI of RNase Po1 is 9.2. At the optimum pH of RNase Po1 and RNase He1, the RNase activity of RNase Po1 was strongly inhibited by Zn2+, similar to that of RNase T1; however, RNase He1 was not inhibited by Zn2+.6,7)
We previously determined the X-ray crystal structure of RNase Po1,8) based on which we postulated that its antitumor activity could result from its high stability because of the presence of disulfide bonds and an overall positive charge on its surface. In this study, we determined the X-ray crystal structure of RNase He1 and investigated the relationship between its structure and antitumor activity by comparing it with the structure of RNase Po1.
MATERIALS AND METHODS
Enzyme Expression and PurificationRNase He1 was expressed in Escherichia coli. The pelB signal sequence from the pET22b expression vector and the He1 gene5) were ligated into the pET11d expression vector (Novagene, Darmstadt, Germany) following the procedure of Huang et al.9) and then transferred to E. coli BL21(DE3)pLysS (Novagene). The transformed E.coli were cultured in Terrific Broth at 25°C for 7 d, with the addition of a final concentration of 100 µg/mL of ampicillin and 0.5 mM isopropyl β-D-1-thiogalactopyranoside (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). The E. coli culture was centrifuged at 12000 × g for 30 min. Since He1 is secreted from the E. coli, the supernatant of the culture was used for subsequent purification by a previously reported method.5) The supernatant was precipitated with ammonium sulfate (90% saturation) and subjected to gel filtration chromatography with column of Sephadex G50 (3 × 180 cm, Ge Healthcare, Uppsala, Sweden) at pH 6.0. The RNase-active fractions of He1 were purified using column chromatography in the following order: Sp-Toyopearl column (3 × 30 cm, TOSOH, Tokyo, Japan) at pH 6.0, DEAE-Toyopearl column (1.5 × 40 cm, TOSOH) at pH 7.5), Ultorogel AcA54 column (3 × 180 cm, Ge Healthcare) at pH 6.0, and heparin-Sepharose column (1 × 20 cm, Ge Healthcare) at pH 4.5. Finally, the RNase-active fractions of He1 were loaded onto an HPLC (SHIMADZU, Kyoto, Japan), LC6A system using a Shodex Protein KW-802.5 column (0.8 × 80 cm, SHOWA DENKO, Nagoya, Japan) at pH 6.0. The RNase-active fractions were pooled as the purified enzyme.
Enzyme AssayRNase activity was measured as described previously using yeast RNA (Marine Biochemicals, Tokyo, Japan) as the substrate at pH 4.5 and 37°C.10)
The experiment for confirming the effect of Zn2+ on RNase activity was performed as follows. ZnCl2 was added to give a final concentration of 1 mM into 0.25% RNA as substrate in 0.05 M Tris–HCl buffer (pH 7.0) or 0.05 M acetate buffer pH 4.5. RNase activity was measured at 37°C as described above.
Protein ConcentrationThe protein concentration of the final purified enzyme was determined spectrophotometrically, assuming an absorbance of 9674 for a one mol/L solution at 280 nm. This value was estimated from the amino acid sequence of RNase He1 (data not shown).
Tricine Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE)Tricine SDS-PAGE was performed using a 15% polyacrylamide gel by Schagger’s method.11) Proteins on the gel were visualized by silver staining. Activity staining of the RNases was conducted using the method of Blank et al.12)
Protein Crystallization and Structural AnalysisCrystallization screening was initially performed using CrystalScreen, CrystalScreen2, Index, polyethylene glycol (PEG)/Ion, and PEGRx (Hampton Research, U.S.A.). A Mosquito LCP (TTP Labtech, U.S.A.) was used for the preparation of screening plates. The protein solution (0.2 µL, 30 mg/mL He1 in 50 mM MES pH 6.5, 50 mM NaCl) was mixed with 0.2 µL reservoir solution. Each drop was equilibrated against 50 µL of reservoir solution using a sitting-drop vapor diffusion method. Initially, we obtained a crystal in CrystalScreen2 #27 (25% (v/v) PEG-MME 550, 0.1 M MES pH 6.5, 10 mM zinc sulfate heptahydrate). Further optimization was carried out around this condition using a 24-well crystallization plate (0.5 µL of protein solution described above was mixed with 0.5 µL of reservoir solution and equilibrated against 500 µL of reservoir solution using hanging-drop vapor diffusion). Finally, we obtained a crystal using the macro seeding method. Needle-shaped crystals obtained from the final condition (31% (v/v) PEG-MME 550, 0.1 M MES pH 7, 0.5 mM zinc sulfate) were crushed and added to newly prepared drops with the same crystallization conditions. No additional cryoprotectant was used for X-ray diffraction experiments and the crystals were flash-cooled in liquid nitrogen. A crystallographic reflection data set was collected on the BL-17A beamline at the Photon Factory (Tsukuba, Japan). The data set was collected under a cryostream at 100 K and 0.9800 Å wavelength with an oscillation angle of 180°, 1° per frame. Indexing and scaling procedures were performed using HKL2000 (HKL Research, U.S.A.). Molecular replacement for phase determination was performed using MOLREP.13) RNase Po1 (PDB ID: 3WHO) was used as the model for molecular replacement. Structure refinement was carried out using Refmac5 and PHENIX14) after manual rebuilding with COOT.15)
Identification of Molecular ZnWe verified the identity of zinc ions with X-ray absorption fine structure (XAFS) spectroscopy and an anomalous difference map was collected at the absorption edge of zinc.
RESULTS
Purification of RNase He1E. coli expressing recombinant RNase He1 secreted approximately 35000 units (approximately 17 mg) of RNase He1 into 2 L of culture supernatant. RNase He1 was purified to homogeneity, as determined by SDS-PAGE (Fig. 2).
Data Collection and Structure DeterminationPurified RNase He1 crystallized in the space group P21 21 21 with unit cell dimensions of a = 31.701 (Å), b = 42.945 (Å), and c = 60.183 (Å). The structure of RNase He1 was solved using molecular replacement and refined to a resolution of 0.98 Å (Table 1).
Table 1. Data Collection and Refinement Statistics Parameters
| Parameter | RNase He1 |
|---|
| Data collection | |
| Beamline | Photon Factory BL-17A |
| Wavelength [Å] | 0.9800 |
| Resolution [Å] | 1.57–34.96 |
| Space group | P21 21 21 |
| Unit cell dimension length [Å] | a = 31.701, b = 42.945, c = 60.183 |
| Angle (°) | α = 90.00 β = 90.00 γ = 90.00 |
| Redundancy | 6.4 |
| Refinement | |
| Resolution [Å] | 1.5–34.96 |
| Number of reflections | 13700 |
| Rwork/Rfree | 0.1725/0.1973 |
| R.m.s deviation | |
| Bond length [Å] | 0.011 |
| Bond angle (°) | 1.24 |
The atomic structure of RNase He1 was very similar to that of RNase T1 and RNase Po1. RNase He1 was an (α + β)-type structure consisting of an α-helix with 3.5 turns (residues 14–25) and seven β-strands (residues 5–7, 10–12, 34–36, 51–55, 70–75, 81–85, 96–97). In RNase He1, the helix ran like a “backbone” down the molecule with a four-stranded anti-parallel β-sheet (34–35, 52–55, 71–75, 81–87) crossing the α-helix, similar to RNase Po1 (Figs. 3a, b). Electron density was not available for residues glutamic acid (Glu)40–Ser43, Ala88–Glu91, Pro45, and Ser46. From this and by comparing the RNase He1 structure with the structures of RNase Po1 and RNase T1, we suggest that these residues constitute flexible loop regions.
The catalytic site of RNase T1 consists of histidine (His)40 and/or Glu58, arginine (Arg)77, and His92.16) Moreover, Steyaert et al.17) and Nonaka et al.18) reported that Glu58, rather than His40, must be the general base catalyst of the RNase T1 family. It has also been reported for RNase T1 that in the transesterification step of phosphodiester hydrolysis, His40 and/or Glu58 act as a general base toward the ribose 2′-hydroxyl group and that His92, as a general acid, donates a proton to the leaving 5′-hydroxyl group.16) The catalytically active amino acid residues of RNase He1 are His34, Glu53, Arg71, and His86, corresponding to RNase T1 His40, Glu58, Arg77, and His92, and RNase Po1 His36, Glu54, Arg72, and His87, respectively. These amino acid residues of RNase T1, RNase Po1, and RNase He1 are located in the β3–6 strands (His34, Glu53, Arg71) and next to the β6 strand (His86) (Fig. 3c).
The base recognition site of RNase T1 reportedly consists of tyrosine (Tyr)42, asparagine (Asn)43, Asn44, Tyr45, Glu46, and Asn98, which are located in the loop between the β3–4 strands (Asn43, Asn44, Tyr45, Glu46) and in the loop between the β6–7 strands (Asn98). We previously reported that RNasePo1 has a base recognition site equivalent to that of RNase T1.
The base recognition site of RNasePo1 consists of Tyr38, Asn39, Asn40, phenylalanine (Phe)41, Glu42, and Asn94, which are equivalent to Tyr36, His37, aspartic acid (Asp)38, Tyr39, Glu40, and Asp93 of RNase He1, respectively. These amino acid residues are also located in the loop between the β3–4 strands (His37, Asp38, Tyr39, Glu40) and in the loop between the β6–7 strands (Asn93). The two aromatic rings of Tyr32 (in the β3 strand) and Tyr39 in RNase He1 may stack with the guanine base. Comparing the base recognition sites between RNase He1, RNase Po1, and RNase T1, an Asn residue is replaced by a His residue and two Asn residues become Asp in RNase He1. As RNase He1 is a guanine base-specific RNase similar to RNase Po1 and RNase T1, these amino acids clearly do not influence base recognition; however, they may play a role in the optimal activity of RNase He1 at pH 4.5. To elucidate the role played by these residues, we need to investigate the structure of RNase He1 bound to its substrate (Fig. 4).
Two important roles for the β-sheets in RNase T1 have been reported,2) and we have previously reported similar roles in RNase Po1.8) First, the β-sheets form an internal hydrophobic core and second, the β-sheet structure forms the catalytic pocket of the enzyme. The three hydrophilic side chains of Glu58, Arg77, and His92 in RNase T1 form a cluster on the β-sheet surface opposite the α-helix and are involved in enzymatic activity. The Glu53 (in the β4-strand), Arg71 (in the β5-strand), and His86 (next to the β6-strand) residues of RNase He1 are conserved within RNase T1 and RNase Po1. The β4–6 sheet of RNase He1 is within the molecule and consists of 16 amino acid residues, seven of which are hydrophobic, as in RNase Po1 (eight hydrophobic residues, 16 total) and RNase T1 (9 hydrophobic residues, 17 total). The hydrophobic amino acid residue clusters of the β4–6 sheet of RNase He1 are opposite the α-helix and may interact with residues from other strands and the α-helix, which then form an internal hydrophobic core similar to RNase T1 and RNase Po1 (Fig. 5).
RNase He1 has six cysteine residues. We determined the disulfide bond combinations of these residues to be Cys7–Cys98, Cys5–Cys83, and Cys47–Cys81 (Figs. 3a, b). The cysteine residues that formed the disulfide bonds of RNase He1 are conserved with RNase Po1.
Identification of Molecular ZnWe confirmed the presence of zinc using XAFS spectroscopy and an anomalous difference map collected at the absorption edge of zinc (Fig. 6a). We collected data for each part of the spectrum with energies converted to wavelength, peak (1.2816 Å), edge (1.2827 Å), remote L (1.2900 Å), and remote H (1.2567 Å), by multiple wavelength anomalous dispersion.19,20) At the peak, we found electron density for molecular Zn, which disappeared at the remote L region. We found that the structure of RNase He1 has two Zn molecules bound. One Zn interacts with Asp75, is bound at the bottom of the RNase He1 molecule, and most likely does not influence enzyme activity (Figs. 6b, d). The other Zn is bound within the molecule interacting with His34, His37, and Glu53 (Figs. 6b, d, e). Furthermore, the Zn is bound within the active pocket of RNase He1 that forms the internal hydrophobic core (Fig. 5).
Effect of Zn2+ on the Activity of RNase He1 and RNase Po1We examined the effect of Zn2+ on RNase activity at pH 4.5 and 7.0. At pH 4.5, neither RNase He1 nor RNase Po1 was inhibited by 1 mM Zn2+. However, at pH 7.0, the remaining activity for both was approximately 5%. Therefore, both enzymes are significantly inhibited by 1 mM Zn2+ at pH 7.0 (Fig. 7).
DISCUSSION
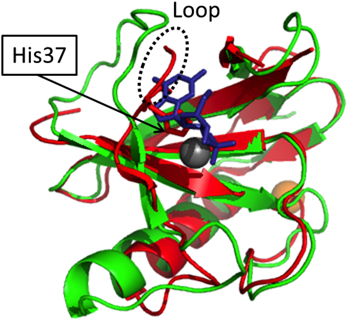
We crystalized RNase He1 at pH 7.0, allowing us to investigate the binding of molecular Zn. One Zn is bound within the enzyme molecule, interacting with His34, His37, and Glu53 of RNase He1. Furthermore, the Zn is within the active pocket of RNase He1, which forms an internal hydrophobic core (Figs. 5, 6). We superimposed the X-ray structure of RNase He1-Zn with that of RNase T1–2′GMP. His37 and Glu53 are conserved with His40 and Glu58 of RNase T1 and are within the active site. The loop containing His37 is in a different region than in RNase T1 because of its interaction with Zn; therefore, this loop may prevent guanine substrate binding due to steric hindrance (Fig. 8), rendering RNase He1 inactive. This should be clarified when the three-dimensional X-ray crystallographic structure of the RNase He1-GMP complex is determined.
The RNase activity of RNase He1 is inhibited by Zn2+ at approximately pH 7.0, similar to RNase Po1 and RNase T1 (Fig. 7); therefore, it is likely that steric hindrance caused by zinc also plays a role in RNase T1 and RNase Po1 inhibition. However, at pH 4.5, molecular Zn may not be able to bind RNase He1, as its enzymatic activity is not inhibited by Zn2+ at its optimum pH. Additionally, because the loop region of Glu40–Ser46 is adjacent to His37, this loop might be affected by Zn and is a random structure.
RNase He1 has high sequence identity with RNase Po1 (60%), their catalytic sites and six cysteine residues involved in disulfide bond formation are conserved, and their X-ray crystal structures are almost identical. We previously reported that the optimum temperature of RNase He1 activity was 20°C higher than that of RNase T1, and similar to that of RNase Po1.4,5) We also reported that a number of Cys residues were responsible for the heat stability of RNase He1.5) One disulfide bond in RNase He1 (Cys7–Cys98) is conserved in all known RNase T1 family members, except those of bacterial origin. Using mutagenesis, we determined that the Cys7–Cys98 and Cys5–Cys83 bonds were structurally essential for the RNase active site while Cys47–Cys81 helped maintain conformational stability.5)
After observing the X-ray crystal structure of RNase He1 in the present study, we confirmed the location of the three disulfide bonds. The Cys5–Cys83 bond is located parallel to the Cys7–Cys98 bond, making the C- and N-termini of RNase He1 more rigid than RNase T1. The Cys47–Cys81 bond of RNase He1 is located near the active site on the opposite side of the Cys5–Cys83 bond. Moreover, the Cys47–Cys81 bond connects the β6-strand to the loop between the β3- and β4-strands. The disulfide bond connecting to the flexible loop may be less important for stability than the other disulfide bonds. However, there are amino acid residues (Tyr36, Asp38, Tyr39, and Glu40) within this loop that are believed to constitute the base recognition site. Next to the β6-strand, there is one catalytic residue (His87). Therefore, the Cys5–Cys83 and Cys47–Cys81 bonds stabilize the catalytic pocket and may contribute to the higher heat stability of RNase He1. We conclude that the higher heat stabilities of RNase Po1 and RNase He1 in comparison to that of RNase T1 are due to the stabilizing effect of the additional disulfide bonds (Fig. 3). The RNase specific activity of RNase He1, RNase Po1, and RNase T1 for degrading RNA are almost same.1,3,5) Thus, it seems that the difference in the stabilizing effect had little influence on RNase activity. We previously reported that RNase Po1 is more resistant to chymotrypsin- and thermolysin-mediated proteolysis than RNase T1, and that RNase Po1 inhibits the proliferation in HL-60 cells more than RNase T1 after transfection using a cell-penetrating peptide.8) Therefore, the abovementioned stabilizing effect might be more important within the tumor cell.
Cytotoxic RNases attack intracellular RNA and must be internalized.21) Johnson et al.22) suggested that a family of cationic onconases of the RNase A family from Rana pipiens bind to the plasma membrane via electrostatic interactions and that this binding is critical to their strong anti-tumor properties.23) Furthermore, it was reported that the membrane surface of many cancer cells is more negatively charged than the membrane surface of normal cells, which is mainly due to an increased number of non-reducing terminal sialic acids in the cell surface carbohydrates.24,25)
Therefore, we also compared the surface electrostatic potentials of RNase He1 and RNase Po126) and found that the surface of RNase Po1 is positively charged while the surface of RNase He1 is negatively charged, especially in the region behind the catalytic site (Fig. 9). RNase He1 has five Arg residues, fewer than RNase Po1, which has eight. Four of the five Arg residues in RNase He1 are located on the surface of the molecule (except for Arg71, which is in the active site), and five Arg residues are located on the surface of RNase Po1. Therefore, in this respect, there is little difference between the two enzymes. However, RNase He1 has nine Asp and seven Glu residues, while RNase Po1 has only three of each. All of the Asp and Glu residues in RNase He1 are surface exposed, except for Glu53, which is located in the active site. However, only two Glu residues are surface exposed in RNase Po1. Therefore, the exterior Asp and Glu residues in RNase He1, in contrast to RNase Po1, are responsible for the negatively charged surface of the enzyme.
Regarding the effect of these surface residues on protein activity, we previously reported the mutagenesis of RNase He1, in which seven Asp residues were mutated to Asn and five Glu residues were mutated to Gln. The RNase He1 mutant exhibited anti-tumor activity similar to that of RNase Po1.6) When we refined the X-ray crystal structure of RNase He1, we observed that these residues were surface exposed and the surface electrostatic potential of mutated RNase He1 was positive. In contrast, the optimum pH of RNase He1 is 4.5; therefore, the enzymatic activity of RNase He1 is lower than that of RNase Po1 (optimum pH of 7.5) in the tumor cell line (pH 7.0). However, onconase exhibits low levels of RNase activity27) but possesses strong antitumor activity. Therefore, once the mutant RNase He1 enters the cell, it could exhibit anti-tumor activity despite its low activity.
In conclusion, we investigated the X-ray crystal structure of RNase He1 bound to molecules of Zn, with one located in the active pocket causing a conformational shift in the loop, which likely prevents substrate insertion due to steric hindrance, rendering the enzyme inactive. This may be the mechanism of how Zn inactivates RNase T1 family enzymes. The X-ray crystal structure of RNase He1 was very similar to that of RNase Po1, the latter of which exhibits antitumor activity, with the active site and disulfide bonds responsible for high stability retained between the two enzymes. However, RNase He1 did not have anti-tumor activity, most likely owing to its altered surface electrostatic potential due to Asn to Asp and Glu to Gln conversions. The surfaces of tumor cells are more anionic than normal cells; therefore, when the RNase enters the cell, a cationic enzyme will be more advantageous than an anionic one. The structure of RNase He1 is similar to that of RNase Po1; therefore, it could be a good model for investigating the anti-tumor activity of RNase Po1 toward human tumor cells. Further investigations into the relationship between the structure and anti-tumor activity of RNase enzymes may lead to the development of new antitumor drugs.
Acknowledgments
We thank Dr. Ryuichi Kato for the crystallization of RNase He1.
This research is (partially) supported by the Platform Project for Supporting in Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and the Japan Agency for Medical Research and Development (AMED) (Proposal No. 2012G001, 2013R-11). This work was performed under the approval of the Photon Factory Program Advisory Committee (Proposal No. 2017G148). This work was performed in part under the Collaborative Research Program of Institute for Protein Research, Osaka University, CR-15-05, CR-16-05, CR-17-05, CR-18-05. The atomic coordinates and structure factors (PDB ID code 5GY6) have been deposited in the Worldwide Protein Date Bank (http://www.wwpdb.org/)
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Yoshida H. The ribonuclease T1 family. Methods in Enzymology. (Nicolson AW ed.) Vol. 341, Academic Press, New York, pp. 28–41 (2001).
- 2) Takahashi K. The amino acid sequence of ribonuclease T-1. J. Biol. Chem., 240, 4117–4119 (1965).
- 3) Nomura H, Inokuchi N, Kobayashi H, Koyama T, Iwama M, Ohgi K, Irie M. Purification and primary structure of a new guanylic acid specific ribonuclease from Pleurotus ostreatus. J. Biochem., 116, 26–33 (1994).
- 4) Kobayashi H, Motoyoshi N, Itagaki T, Tabata K, Suzuki T, Inokuchi N. The inhibition of human tumor cell proliferation by RNase Pol, a member of the RNase T1 family, from Pleurotus ostreatus. Biosci. Biotechnol. Biochem., 77, 1486–1491 (2013).
- 5) Kobayashi H, Motoyoshi N, Itagaki T, Inokuchi N. Mutagenesis of the novel Hericium erinaceus Ribonuclease, RNase He1, reveals critical responsible residues for enzyme stability and activity. Biol. Pharm. Bull., 37, 1843–1847 (2014).
- 6) Kobayashi H, Motoyoshi N, Itagaki T, Suzuki M, Inokuchi N. Effect of the replacement of aspartic acid/glutamic acid residues with asparagine/glutamine residues in RNase He1 from Hericium erinaceus on inhibition of human leukemia cell line proliferation. Biosci. Biotechnol. Biochem., 79, 211–217 (2015).
- 7) Uchida T, Egami F. Microbial ribonucleases with special reference to RNases T1, T2, N1, and U2. The Enzymes. (Boyer PD ed.) Vol. 4, Academic Press, New York. pp. 205–225 (1965).
- 8) Kobayashi H, Katsutani T, Hara Y, Motoyoshi N, Itagaki T, Akita F, Higashiura A, Yamada Y, Inokuchi N, Suzuki M. X-ray crystallographic structure of RNase Po1 that exhibits anti-tumor activity. Biol. Pharm. Bull., 37, 968–978 (2014).
- 9) Huang HC, Wang SC, Leu YJ, Lu SC, Liao YD. The Rana catesbeiana rcr gene encoding a cytotoxic ribonuclease. Tissue distribution, cloning, purification, cytotoxicity, and active residues for RNase activity. J. Biol. Chem., 273, 6395–6401 (1998).
- 10) Irie M. Isolation and properties of a ribonuclease from Aspergillus saitoi. J. Biochem., 62, 509–518 (1967).
- 11) Schägger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem., 166, 368–379 (1987).
- 12) Blank A, Sugiyama R, Dekker CA. Activity staining of nucleolytic enzymes after sodium dodecyl sulfate-polyacrylamide gel electrophoresis: use of aqueous isopropanol to remove detergent from gels. Anal. Biochem., 120, 267–275 (1982).
- 13) Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst., 30, 1022–1025 (1997).
- 14) Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr., 66, 213–221 (2010).
- 15) Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr., 60, 2126–2132 (2004).
- 16) Arni R, Heinemann U, Tokuoka R, Saenger W. Three-dimensional structure of the ribonuclease T1/2-GMP complex at 1.9-A resolution. J. Biol. Chem., 263, 15358–15368 (1988).
- 17) Steyaert J, Hallenga K, Wyns L, Stanssens P. Histidine-40 of ribonuclease T1 acts as base catalyst when the true catalytic base, glutamic acid-58, is replaced by alanine. Biochemistry, 29, 9064–9072 (1990).
- 18) Nonaka T, Nakamura KT, Uesugi S, Ikehara M, Irie M, Mitsui Y. Crystal structure of ribonuclease Ms (as a ribonuclease T1 homologue) complexed with a guanylyl-3′,5′-cytidine analogue. Biochemistry, 32, 11825–11837 (1993).
- 19) Hendrickson WA. Determination of macromolecular structures from anomalous diffraction of synchrotron radiation. Science, 254, 51–58 (1991).
- 20) Hendrickson WA, Ogata CM. Phase determination from multiwavelength anomalous diffraction measurements. Methods Enzymol., 276, 494–523 (1997).
- 21) Wu Y, Mikulski SM, Ardelt W, Rybak SW, Youle RJ. A cytotoxic ribonuclease. Study of the mechanism of onconase cytotoxicity. J. Biol. Chem., 268, 10686–10693 (1993).
- 22) Johnson RJ, Chao TY, Lavis LD, Raines RT. Cytotoxic ribonucleases: the dichotomy of Coulombic forces. Biochemistry, 46, 10308–10316 (2007).
- 23) Darzynkiewicz Z, Carter SP, Mikulski SM, Ardelt W, Shogen K. Cytostatic and cytotoxic effects of Pannon (P-30 Protein), a novel anticancer agent. Cell Tissue Kinet., 21, 169–182 (1988).
- 24) Kobata A, Amano J. Altered glycosylation of proteins produced by malignant cells, and application for the diagnosis and immunotherapy of tumours. Immunol. Cell Biol., 83, 429–439 (2005).
- 25) Lau KS, Dennis JW. N-Glycans in cancer progression. Glycobiology, 18, 750–760 (2008).
- 26) Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A., 98, 10037–10041 (2001).
- 27) Ardelt W, Mikulski SM, Shogen K. Amino acid sequence of an anti-tumor protein from Rana pipiens oocytes and early embryos. Homology to pancreatic ribonucleases. J. Biol. Chem., 266, 245–251 (1991).