Abstract
Pain is a defense system that responds rapidly to harmful internal and external stimuli through the somatosensory neuronal pathway. However, damage to the nervous system through cancer, diabetes, infection, autoimmune disease, chemotherapy or trauma often leads to neuropathic pain, a debilitating chronic pain condition. Neuropathic pain is not simply a temporal continuum of acute nociceptive signals from the periphery, but rather due to pathologically altered functions in the nervous system, which shift the net neuronal excitatory balance toward excitation. Although alterations were long thought to be a result of changes in neurons, but an increasing body of evidence over the past decades indicates the necessity and sufficiency of microglia, the tissue-resident macrophages of the spinal cord and brain, for nerve injury-induced malfunction of the nervous system. In this review article, I describe our current understanding of the molecular and cellular mechanisms underlying the role of microglia in the pathogenesis of neuropathic pain and discuss the therapeutic potential of microglia from recent advances in the development of new drugs targeting microglia.
1. INTRODUCTION
Human and other organisms acquire nociceptive pain as a defense system that rapidly responds to a wide range of harmful internal and external stimuli that threaten physiological homeostasis. Noxious stimuli such as heat and mechanical and chemical irritants to the skin excite nociceptive primary afferent sensory neurons, called nociceptors (thin, myelinated Aδ and unmyelinated C fibers). The excitatory information is then conveyed to the spinal dorsal horn (SDH) and activates brain-projecting nociceptive pain transmission neurons in lamina I1–4) via complex neuronal circuits. The information then goes up to the brainstem and higher brain regions related to sensory and affective components of pain.1,5–7) In contrast, somatosensory signals evoked by innocuous stimuli such as touch to the skin are also conveyed to the SDH through low threshold mechanoreceptors (LTMRs) including thick, myelinated Aβ fibers, which activate distinct SDH neuronal circuits. These Aβ fibers anatomically connect to nociceptive lamina I neurons via excitatory and inhibitory interneurons, but the signaling flow from Aβ fibers is normally interrupted by inhibitory synaptic inputs from γ-aminobutyric acid (GABA)- or glycine-containing interneurons. Thus, under normal healthy conditions, Aβ fibers do not activate nociceptive projection neurons and do not cause pain. As such, the accurate perception of pain, as well as other sensations, requires properly wired neuronal circuits and their correct function.
However, following damage to the nervous system such as by cancer, diabetes, infection, autoimmune disease, chemotherapy and trauma, this system can malfunction. These conditions often cause a debilitating chronic pain syndrome (termed neuropathic pain). A cardinal symptom of neuropathic pain is mechanical allodynia (touch-evoked pain). Neuropathic pain does not resolve even after the healing of tissue damage, and can persist for long periods of time. Thus, it is considered that the pain is not simply a temporal continuum of acute nociceptive signals from the periphery, but rather due to pathologically altered functions in the nervous system.2,4,8,9) Pathological changes and their molecular and cellular mechanisms have been studied using models of neuropathic pain, for example rodent models established by peripheral nerve injury (PNI). A growing body of evidence indicates that PNI causes anatomical and functional modifications in primary afferent sensory neurons, SDH neurons and the brain. These modifications shift the balance between synaptic excitation and inhibition toward excitation, which may account for development and maintenance of chronic pain.2,4,8,9) These alterations were long thought to be a consequence simply of changes in neurons, but accumulating evidence during the last 15 years indicates the important role of non-neuronal cells of the nervous system, including monocytes, macrophages, T cells, and glial cells.10,11) In this review article, I describe the molecular and cellular mechanisms underlying the role of microglia in the pathogenesis of neuropathic pain and also discuss the therapeutic potential of microglia from recent advances in the development of new drugs targeting microglia.
2. MICROGLIA
Microglial cells are known as the tissue-resident macrophages of the central nervous system (CNS) and constitute 5–10% of total cells in the adult CNS. Microglia were originally described by Pio del Rio-Hortega in 191912) and proposed to have a mesodermal origin.13) By fate mapping analyses, erythromyeloid progenitors generated in the embryonic yolk sac were identified as the origin of microglia.14) The progenitors develop into microglia progenitors via an immature and more mature stage, then leave the yolk sac and migrate to the brain through blood vessels, and, terminally differentiate into microglia. In the healthy CNS, microglia remain throughout life and are maintained by self-renewal.15) As microglia have a unique molecular signature compared with other myeloid and immune cells such as circulating monocytes or macrophages,16–19) a distinct gene expression program is required for the development of microglia. It has been shown that interleukin-34 (IL-34) is crucial for their development20,21) and transforming growth factor-β1 (TGF-β1) is required for promoting terminal differentiation and acquiring adult microglia properties.17) For maintaining microglia in adults, colony stimulating factor 1 receptor (CSF1R) signaling might have an ongoing role.22) Microglia in the adult CNS represent a unique morphology, which has a small soma with thin and branched processes whose motility is highly dynamic.23–25) Furthermore, microglia directly contact presynaptic terminals and dendritic spines and, in response to neuronal excitation, extend their processes toward highly active synapses.26) Now, microglia in the CNS are received much attention as being crucial for forming and refining neuronal circuitry and network connectivity, and contributing to neuronal plasticity.27)
3. MICROGLIOSIS IN THE SPINAL DORSAL HORN AFTER PNI
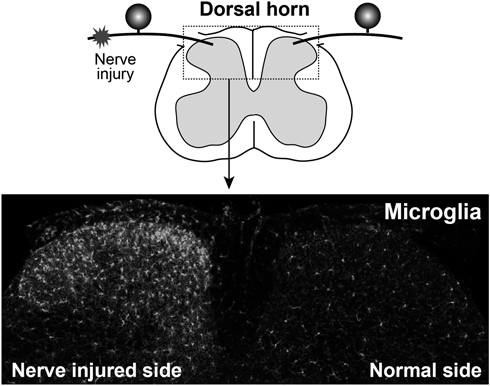
Injury to the peripheral nerves activates microglia in the SDH (Fig. 1). In the late 1970s, it was found that non-neuronal cells (which were later identified as microglia) are increased in the SDH after PNI.28,29) Two possibilities have been considered as the mechanisms for the PNI-induced SDH microgliosis. First is proliferation of resident microglia because after PNI, SDH microglia was found to be immunohistochemically labeled by proliferation markers.30) A recent study examining the detailed temporal kinetics of microgliosis by a short-pulse labeling of proliferating cells showed a narrow time window for rapidly inducing a proliferation burst of SDH microglia after PNI.31) One potential candidate factor recently identified is CSF1.32,33) Consistent with the kinetics of microglial proliferation after PNI, CSF1 expression is rapidly induced in injured dorsal root ganglion (DRG) neurons.32,33) This expression seems to involve IL-1β from surrounding satellite glia.34) Conditional knockout of CSF1 in DRG neurons prevented the PNI-induced microglial proliferation, and, conversely, intrathecal administration of CSF1 to normal mice induced proliferation,32) indicating that CSF1 in injured DRG neurons is necessary and sufficient to induce microglial proliferation after PNI.
In contrast, it should be noted that the upregulation of CSF1 is not transient but persists until a few weeks after PNI32,33) when microglial proliferation has already terminated,31,35) suggesting a distinct role for CSF1–CSF1R signaling at this later phase, such as the control of the expression of microglial genes. The second possible mechanism could be infiltration of bone marrow (BM)-derived circulating monocytes into the parenchyma of the SDH, which then differentiate into microglia-like cells.36) This infiltration of BM cells was found in BM chimeric mice that had received lethal irradiation with BM transplantation. The dose of irradiation used was high, which can produce toxic side effects including an induction of chemoattractants and a disruption of the blood brain/spinal cord barrier.37) On the other hand, in BM chimeric mice generated by a lower irradiation dose, such infiltration of BM cells was not observed.38) In addition, several studies also showed no contribution of circulating monocytes to the PNI-induced microgliosis using parabiosis mice (a model in which two mice are surgically joined and share circulating blood)38) and transgenic mice enabling distinct visualization of resident microglia and circulating monocytes.35) Together, it is now appreciated that local expansion of resident microglia by proliferation is the primary cellular mechanism for SDH microgliosis after PNI.31,35,38) Nevertheless, it should be noted that infiltration of circulating monocytes might occur in the SDH of other models of neuropathic pain. For example, in experimental autoimmune encephalomyelitis (EAE; a model of multiple sclerosis, with chronic pain being a common symptom), monocytes massively infiltrated into the spinal cord with demyelinating lesions,39) although these monocytes did not permanently contribute to the resident microglia pool.
4. CAUSAL LINK BETWEEN MICROGLIA AND NEUROPATHIC PAIN
Following initial reports of PNI-induced microgliosis in the SDH,28,29) the role of microglia in neuropathic pain remained unclear for about thirty years. In 2003, a causal link was first uncovered by studies investigating the role of the ionotropic purinergic receptor subtype P2X4 receptor (P2X4R)40) and p38 mitogen-activated protein kinase (p38MAPK).41)
P2X4R is a subtype of the P2X family consisting of ATP-gated ionotropic receptors42,43) that are formed by three subunits44,45) permeable to Na+, K+ and Ca2+.46,47) It was found that expression of P2X4R in the SDH is upregulated exclusively in microglia after PNI, and that pharmacological blockade and genetic knockout of P2X4R suppress the PNI-induced mechanical allodynia,40,48,49) indicating the necessity of microglial P2X4Rs in neuropathic pain. Furthermore, intrathecal administration of P2X4R-stimulated cultured microglia to normal rats induced allodynia,40) indicating its sufficiency.50) The EAE model of multiple sclerosis—with its common symptom of chronic pain51)—showed microglial activation and P2X4R upregulation in the SDH, and mechanical allodynia.52–54) An increase in expression of P2X4R was also found in SDH microglia of a model of acute inflammatory demyelinating polyradiculoneuropathy, the most common subtype of Guillain-Barre syndrome.55) In a model of herpetic pain, microglia-selective upregulation of P2X4R in the SDH was also found, and its time course corresponded with mechanical allodynia.56) The herpes-induced mechanical allodynia was suppressed by pharmacological blockade of P2X4Rs. Thus, microglia activation and their expression of P2X4R are commonly observed phenomena in diverse models of neuropathic pain. However, the contribution of microglial P2X4R in neuropathic pain may be different between males and females. It was reported that PNI did not upregulate P2X4R expression and that pharmacological inhibition of P2X4R had no effect on mechanical allodynia57) in female mice. A sex difference in the role of microglial molecules in neuropathic pain was also reported in Toll-like receptor 4 (TLR4)58) and p38MAPK,59) both of which have a crucial role in spinal microglia in males.41,58–61) Nevertheless, there are also reports showing no obvious sexual dimorphism in the suppressive effect on PNI-induced pain by genetic knockout of microglia-selective genes including the G-protein-coupled P2 receptor P2Y12R62) and C-X3-C motif chemokine receptor 1 (CX3CR1).63) In addition, P2X4R upregulation in spinal microglia of females has been observed in rodent models of bone cancer pain64) and herpetic pain.56) In the latter model, microglial P2X4R activates the brain-derived neurotrophic factor (BDNF)-tropomyosin-related kinase B (TRKB) signaling pathway.56) Thus, further work needs to be performed to clarify the sex-dependent contribution of SDH microglia in other models of neuropathic pain.
5. MOLECULAR MECHANISMS FOR EXPRESSION OF MICROGLIAL P2X4R
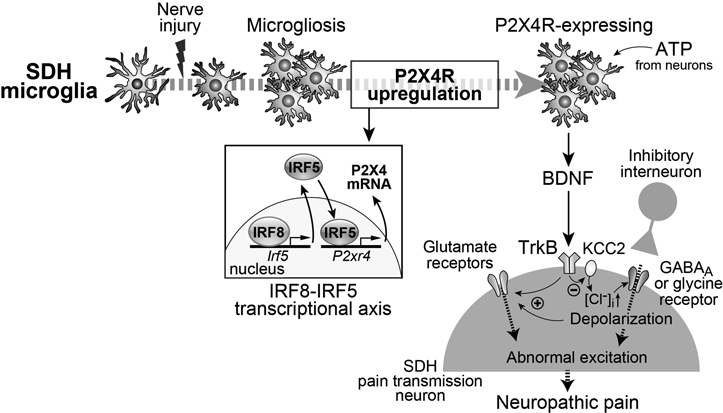
PNI-induced microglial activation dramatically changes expression of genes. Besides P2X4R, numerous microglia-selective molecules (approximately 40) are currently identified as being implicated in PNI-induced pain.11) One of the key regulators which govern a microglia-selective transcription in the context of PNI is interferon regulatory factor 8 (IRF8), a member of the IRF family.65) It was demonstrated that after PNI, expression of IRF8 in the SDH is upregulated exclusively in microglia and IRF8 regulated expression of P2X4R and other microglial genes including P2Y12R, TLR2, and CX3CR1 and diffusible factors (IL-1β, cathepsin S (CatS) and BDNF).66) IRF8 directly regulates other members of the IRF family such as IRF1 and IRF5.67,68) IRF5 was shown to bind to the putative promoter region of P2X4R and enhance its expression.67) Indeed, a deficiency of IRF5 suppressed the PNI-induced spinal P2X4R upregulation and neuropathic allodynia. Therefore, the IRF8–IRF5 transcription axis would be an important mechanism for producing P2X4R-expressing microglia after PNI and neuropathic pain (Fig. 2). A recent study also showed that v-maf avian musculoaponeurotic fibrosarcoma oncogene homolog B (MAFB), which is a basic leucine zipper transcription factor, is also involved in microglial P2X4R upregulation after PNI.69) Microglia-selective knockout of MAFB in adult mice suppresses the PNI-induced mechanical allodynia and a knockdown of MAFB reduces P2X4R. However, spinal MAFB knockdown had no effect on established mechanical allodynia, suggesting that MAFB plays an important role in initiation of P2X4R-expressing microglia and neuropathic pain after PNI.
What extracellular factor(s) triggers P2X4R expression in microglia? Because P2X4R expression in SDH microglia was found unaltered in a chronic pain model associated with peripheral tissue inflammation, microglial P2X4R upregulation might involve factors released from damaged DRG neurons. Possible candidates include CSF132) and cysteine-cysteine chemokine ligand 21 (CCL21),70) whose expressions were found to be increased in injured DRG neurons. CSF1 also increased expression of IRF8 in spinal microglia.32) In addition, other extracellular factors whose source remains to be determined could also be involved in the P2X4R expression in microglia.71–74)
6. CELLS RELEASING ATP TO ACTIVATE MICROGLIA
Extracellular ATP must be needed to activate microglial P2X4Rs. ATP has been reported to be released from primary afferents,75) SDH neurons76) and glial cells.77–79) However, the cell type responsible for releasing ATP within the SDH in the context of PNI was unknown. We recently demonstrated that SDH neurons that express vesicular nucleotide transporter (VNUT,80) also known as SLC17A9, a secretory vesicle protein responsible for storage and release of ATP) are a crucial source of the ATP that causes pain hypersensitivity.81) It was found that PNI increases expression of VNUT and extracellular ATP content within the spinal cord. A deficiency of VNUT attenuated the PNI-induced mechanical allodynia. This was phenocopied by a specific knockout of VNUT in SDH neurons, but not in primary sensory neurons, microglia or astrocytes. Thus, VNUT in SDH neurons is necessary for ATP release that contributes to neuropathic pain.
7. MICROGLIA-DERIVED FACTORS CRUCIAL FOR NEUROPATHIC PAIN
Activated microglia also express a variety of humoral factors such as proinflammatory cytokines, chemokines and trophic factors. After stimulation of P2X4R in microglia, BDNF was found to be released.82,83) BDNF was then shown to act on TrkB in lamina I neurons and to induce an altered transmembrane anion gradient by downregulating K+-Cl− co-transporter 2 (KCC2), which causes changes in GABA- and glycine-evoked responses from inhibitory to excitatory and mechanical hypersensitivity82) (Fig. 2). This abnormal excitation results in potentiating the glutamatergic excitation via N-methyl-D-aspartate receptors (NMDAR).84) The necessity of microglial BDNF for PNI-induced pain was revealed by the data obtained from microglia-selective BDNF deficient mice.57) These findings indicate that the P2X4R–BDNF–KCC2 pathway is a crucial microglia–neuron communication responsible for neuropathic pain (Fig. 2).
Other microglial molecules signaling to SDH neurons that have been extensively studied are the proinflammatory cytokines IL-1β and tumor necrosis factor-α (TNFα).11) Important microglial receptors for producing and releasing these proinflammatory cytokines might be P2X7R and TLRs.60,85–87) In the SDH, P2X7R mediates ATP-induced IL-1β release from TLR4-primed microglia.88) PNI-induced IL-1β transcription in the spinal cord involves TLR287) and TLR4.60) At a post-transcription level, the Nod-like receptor family, pyrin domain containing-3 protein (NLRP3) inflammasomes activate pro-caspase-1, which promotes pro-IL-1β processing and secretion of mature IL-1β.89) IL-1β has been shown to phosphorylate NMDARs90) and to enhance excitatory synaptic transmission.91–93) IL-1β also decreases GABA- and glycine-mediated synaptic inhibition.92) In addition, microglial IL-18 acts on astrocytes, which contributes to neuropathic pain.94) SDH astrocytes also become activated after PNI and contribute to maintenance of pain hypersensitivity,95–97) suggesting a crucial role of microglia–astrocyte signaling in pain chronicity in the context of PNI. As for TNFα, its expression in the SDH was found to be increased in microglia after PNI via p38MAPK.98) In SDH neurons, TNFα rapidly increases excitatory responses evoked by activation of NMDARs and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs).92,99) In microglia, activation of TNF receptors increases expression of BDNF, which was shown to increase dendritic structural remodeling and synaptic connectivity strength in lamina I SDH neurons.100) Collectively, TNFα modulates synaptic structure and strength in SDH neurons by multiple mechanisms involving direct and indirect effects.
CatS is a lysosomal cysteine protease that was found to be released from microglia.101) It was shown that microglial CatS released in response to P2X7R activation via p38MAPK cleaves membrane-bound fractalkine expressed on SDH neurons and astrocytes.102) The cleaved fractalkine is considered to act on microglia again because the fractalkine receptor CX3CR1 is expressed exclusively in microglia.35,103) Activation of the P2X7R–p38MAPK–CatS–fractalkine–CX3CR1 pathway leads to IL-1β secretion from microglia,93) which in turn modulates synaptic excitation and inhibition, as described above.
8. DEVELOPMENT OF MICROGLIA-TARGETING DRUGS
The preclinical studies described indicate great interest in microglia as a promising target for treating neuropathic pain. To date there are no clinically approved drugs that selectively target microglia and their molecules, but drug discovery efforts are currently in progress. As for P2X4Rs, some selective antagonists were developed.104) These include the benzodiazepine derivative 5-BDBD,105,106) the N-substituted phenoxazines PSB-12054 and PSB-12062,107) and the phenylurea BX430.108) The compound NP-1815-PX which was more recently identified as a novel P2X4R antagonist was easily dissolved in water and inhibited rodent and human P2X4Rs with a high potency.56) Among the P2XRs tested, this compound was selective for P2X4R. Most importantly, NP-1815-PX had an anti-allodynic effect in pathological chronic pain models without any alterations in acute physiological pain responses or motor coordination, which are predicted therapeutic benefits of this antagonist. Unfortunately, NP-1815-PX had poor penetration to the CNS tissues, but the pharmaceutical company Nippon Chemiphar successfully developed a more potent and specific P2X4R antagonist with CNS-penetrating properties (NC-2600), which are now under clinical trials in Japan.11) Furthermore, recent studies developed monoclonal antibodies for extracellular head domains of P2X4Rs,109–111) a region that is an important site for modulation of this receptor.45) Among the developed antibodies, immunoglobulin G (IgG)#191-Bbbt0626 produced dose-dependent and long-lasting analgesia (for at least seven days) in a neuropathic pain model when given subcutaneously.111) In addition, a chemical screening of a library identified the antidepressant duloxetine as a clinically approved drug that has an inhibitory effect on rodent and human P2X4R and a reversal effect on the PNI-induced mechanical allodynia.112) For drugs controlling ATP release, the first-generation bisphosphonate clodronate was identified as a potent and selective allosteric inhibitor for VNUT. Clodronate impaired vesicular ATP release from neurons and attenuated neuropathic pain.113) For P2X7R–CatS–fractalkine/CX3CR1–p38MAPK–IL-1β pathway, much effort has been made to develop selective P2X7R antagonists. These include A-740003 dose-dependently reduced nociception in rodent models of neuropathic and inflammatory pain,114) which was lost in IL-1-knockout mice. While several compounds had problems–especially a poor CNS penetration–JNJ-47965567 was identified as a brain-penetrant P2X7R antagonist and had analgesic effects on neuropathic hypersensitivity.115) JNJ-42253432 had a higher CNS penetrating property but, surprisingly, no analgesic effect.116) While mechanical hypersensitivity in a collagen-induced arthritis model is attenuated by intrathecal P2X7R antagonists,117) clinical trials of P2X7 antagonists in rheumatoid arthritis have failed.118) The role of P2X7R is thus still controversial. A selective inhibitor for CatS was also reported to attenuate neuropathic allodynia and to potentiate an anti-allodynic efficacy of pregabalin without influencing its side effects.119) For the complement receptor, complement component fragment 5a receptor (C5aR), which has been implicated in PNI-induced pain,120) a potent and orally active C5aR non-competitive allosteric inhibitor, DF2593A, suppressed pain behaviors in various pain models including neuropathic pain.121)
Discovery of a tool to monitor microglia activity or state in the spinal cord and brain would provide an extremely important step forward to understanding the alteration in microglial functions in neuropathic pain patients. Recently, a technique for developing induced microglia-like (iMG) cells from human blood monocytes was reported.122) The origin of circulating monocytes in adults could be different from brain microglia in humans, but a similar pattern of gene expression was observed between iMG and primary microglia obtained from fetal brain tissue.123) Of particular interest, iMG cells of patients with fibromyalgia were demonstrated to show a TNFα-releasing inflammatory phenotype that correlated with pain severity.124) Although further studies are needed to elucidate the role of iMG cells in chronic pain and their function in patients with other chronic pain conditions, iMG cells may be used to study the mechanisms of neuropathic pain and also as biomarkers for diagnosis and therapeutics.
9. MICROGLIA AND OPIOID TOLERANCE/DEPENDENCE
An alternative benefit of microglia-targeting drugs in pain therapy would be to increase the usefulness of opioids, especially tolerance to their analgesic effect and dependence. Chronic treatment with morphine activates microglia in the SDH and some brain regions125) and depleting spinal microglia126) or inhibiting microglial molecules126–129) suppresses analgesic tolerance to opioids. However, spinal microglia had little role in already established tolerance,130) suggesting a contribution to development but not maintenance of analgesic tolerance to opioids. Furthermore, it was also found that spinal microglia depletion attenuates the behavioral sequelae of withdrawal from chronic morphine.79) Mechanistically, microglia activated by chronic morphine treatment release ATP via pannexin 1 that has interacted with P2X7R, which in turn leads to long-term synaptic facilitation in the SDH and the resultant withdrawal signs.79) From these results, it is suggested that targeting spinal microglia might selectively prevent the undesirable side effects related to chronic use of opioids without reducing their analgesic effect. However, how opioids activate microglia remains controversial. While some studies report that opioids increase expression of microglial molecules (such as P2X4R, P2X7R, and pannexin 1) in primary cultured microglia via μ-opioid receptors (MOR),79,128,130) another study show that MOR is not expressed in spinal cord microglia isolated from adult mice.131) This is an important issue that should be clarified by further investigations.
10. CONCLUSION
Accumulating evidence from basic pain research has not only demonstrated the necessity and sufficiency of spinal cord microglia in the pathogenesis of neuropathic pain, but also greatly advanced our understanding of the mechanisms of this contribution at the molecular and cellular levels. The transcriptome analysis of microglia-selective genes16–19) and microglia heterogeneity132) will accelerate investigations. Furthermore, recent work has also showed a crucial role for brain microglia in somatosensory and/or emotional aspects of neuropathic pain.11) Because pharmacological, molecular, and genetic operations of the function or expression of microglia-expressing molecules strongly influence neuropathic pain behaviors and have no effect on acute pain under normal conditions, microglia and their molecules might be potential targets for treating neuropathic pain. Structure-based drug discovery and technological advances in establishing iMG from patients’ monocytes will allow us to establish a strategy to strongly suppress activated microglia and to diagnose neuropathic pain.
This review of the author’s work was written by the author upon receiving the 2019 Pharmaceutical Society of Japan Award for Divisional Scientific Promotion.
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number 19H05658, by the Core Research for Evolutional Science and Technology (CREST) program from Japan Agency for Medical Research and Development (AMED) under Grant Number 19gm0910006, and Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from AMED under Grant Number 19am0101091.
Conflict of Interest
The author declares no conflict of interest.
REFERENCES
- 1) Todd AJ. Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci., 11, 823–836 (2010).
- 2) Braz J, Solorzano C, Wang X, Basbaum AI. Transmitting pain and itch messages: a contemporary view of the spinal cord circuits that generate gate control. Neuron, 82, 522–536 (2014).
- 3) Prescott SA, Ma Q, De Koninck Y. Normal and abnormal coding of somatosensory stimuli causing pain. Nat. Neurosci., 17, 183–191 (2014).
- 4) Peirs C, Seal RP. Neural circuits for pain: Recent advances and current views. Science, 354, 578–584 (2016).
- 5) Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur. J. Pain, 9, 463–484 (2005).
- 6) Bushnell MC, Ceko M, Low LA. Cognitive and emotional control of pain and its disruption in chronic pain. Nat. Rev. Neurosci., 14, 502–511 (2013).
- 7) Bliss TV, Collingridge GL, Kaang BK, Zhuo M. Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat. Rev. Neurosci., 17, 485–496 (2016).
- 8) Kuner R, Flor H. Structural plasticity and reorganisation in chronic pain. Nat. Rev. Neurosci., 18, 20–30 (2017).
- 9) Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science, 288, 1765–1768 (2000).
- 10) Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov., 13, 533–548 (2014).
- 11) Inoue K, Tsuda M. Microglia in neuropathic pain: cellular and molecular mechanisms and therapeutic potential. Nat. Rev. Neurosci., 19, 138–152 (2018).
- 12) Del Rio-Hortega P. El tercer elemento de los centros nerviosos I La microglia en estado normal II Intervencíon de la microglia en los procesos patológicos III Naturaleza probable de la microglia. Bol. de la Soc. Esp. de boil., 9, 69–120 (1919).
- 13) Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol. Rev., 91, 461–553 (2011).
- 14) Kierdorf K, Masuda T, Jordao MJC, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat. Rev. Neurosci., 20, 547–562 (2019).
- 15) Tay TL, Mai D, Dautzenberg J, Fernandez-Klett F, Lin G, Sagar, Datta M, Drougard A, Stempfl T, Ardura-Fabregat A, Staszewski O, Margineanu A, Sporbert A, Steinmetz LM, Pospisilik JA, Jung S, Priller J, Grün D, Ronneberger O, Prinz M. A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat. Neurosci., 20, 793–803 (2017).
- 16) Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, Helft J, Chow A, Elpek KG, Gordonov S, Mazloom AR, Ma’ayan A, Chua WJ, Hansen TH, Turley SJ, Merad M, Randolph GJ. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol., 13, 1118–1128 (2012).
- 17) Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci., 17, 131–143 (2014).
- 18) Gosselin D, Link VM, Romanoski CE, Fonseca GJ, Eichenfield DZ, Spann NJ, Stender JD, Chun HB, Garner H, Geissmann F, Glass CK. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell, 159, 1327–1340 (2014).
- 19) Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci., 16, 1896–1905 (2013).
- 20) Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330, 841–845 (2010).
- 21) Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS, Colonna M. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol., 13, 753–760 (2012).
- 22) Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL, Green KN. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82, 380–397 (2014).
- 23) Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci., 8, 752–758 (2005).
- 24) Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science, 308, 1314–1318 (2005).
- 25) Davalos D, Akassoglou K. In vivo imaging of the mouse spinal cord using two-photon microscopy. J. Vis. Exp., 2012, e2760 (2012).
- 26) Wake H, Moorhouse AJ, Miyamoto A, Nabekura J. Microglia: actively surveying and shaping neuronal circuit structure and function. Trends Neurosci., 36, 209–217 (2013).
- 27) Wilton DK, Dissing-Olesen L, Stevens B. Neuron-glia signaling in synapse elimination. Annu. Rev. Neurosci., 42, 107–127 (2019).
- 28) Gilmore SA. Proliferation of non-neuronal cells in spinal cords of irradiated, immature rats following transection of the sciatic nerve. Anat. Rec., 181, 799–811 (1975).
- 29) Gilmore SA, Skinner RD. Intraspinal non-neuronal cellular responses to peripheral nerve injury. Anat. Rec., 194, 369–387 (1979).
- 30) Gehrmann J, Banati RB. Microglial turnover in the injured CNS: activated microglia undergo delayed DNA fragmentation following peripheral nerve injury. J. Neuropathol. Exp. Neurol., 54, 680–688 (1995).
- 31) Kohno K, Kitano J, Kohro Y, Tozaki-Saitoh H, Inoue K, Tsuda M. Temporal kinetics of microgliosis in the spinal dorsal horn after peripheral nerve injury in rodents. Biol. Pharm. Bull., 41, 1096–1102 (2018).
- 32) Guan Z, Kuhn JA, Wang X, Colquitt B, Solorzano C, Vaman S, Guan AK, Evans-Reinsch Z, Braz J, Devor M, Abboud-Werner SL, Lanier LL, Lomvardas S, Basbaum AI. Injured sensory neuron-derived CSF1 induces microglial proliferation and DAP12-dependent pain. Nat. Neurosci., 19, 94–101 (2016).
- 33) Okubo M, Yamanaka H, Kobayashi K, Dai Y, Kanda H, Yagi H, Noguchi K. Macrophage-colony stimulating factor derived from injured primary afferent induces proliferation of spinal microglia and neuropathic pain in rats. PLOS ONE, 11, e0153375 (2016).
- 34) Lim H, Lee H, Noh K, Lee SJ. IKK/NF-kappaB-dependent satellite glia activation induces spinal cord microglia activation and neuropathic pain after nerve injury. Pain, 158, 1666–1677 (2017).
- 35) Gu N, Peng J, Murugan M, Wang X, Eyo UB, Sun D, Ren Y, DiCicco-Bloom E, Young W, Dong H, Wu LJ. Spinal microgliosis due to resident microglial proliferation is required for pain hypersensitivity after peripheral nerve injury. Cell Reports, 16, 605–614 (2016).
- 36) Zhang J, Shi XQ, Echeverry S, Mogil JS, De Koninck Y, Rivest S. Expression of CCR2 in both resident and bone marrow-derived microglia plays a critical role in neuropathic pain. J. Neurosci., 27, 12396–12406 (2007).
- 37) Larochelle A, Bellavance MA, Michaud JP, Rivest S. Bone marrow-derived macrophages and the CNS: an update on the use of experimental chimeric mouse models and bone marrow transplantation in neurological disorders. Biochim. Biophys. Acta, 1862, 310–322 (2016).
- 38) Tashima R, Mikuriya S, Tomiyama D, Shiratori-Hayashi M, Yamashita T, Kohro Y, Tozaki-Saitoh H, Inoue K, Tsuda M. Bone marrow-derived cells in the population of spinal microglia after peripheral nerve injury. Sci. Rep., 6, 23701 (2016).
- 39) Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci., 14, 1142–1149 (2011).
- 40) Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature, 424, 778–783 (2003).
- 41) Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J. Neurosci., 23, 4017–4022 (2003).
- 42) Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev., 87, 659–797 (2007).
- 43) Khakh BS, North RA. Neuromodulation by extracellular ATP and P2X receptors in the CNS. Neuron, 76, 51–69 (2012).
- 44) Shinozaki Y, Sumitomo K, Tsuda M, Koizumi S, Inoue K, Torimitsu K. Direct observation of ATP-induced conformational changes in single P2X4 receptors. PLoS Biol., 7, e1000103 (2009).
- 45) Kawate T, Michel JC, Birdsong WT, Gouaux E. Crystal structure of the ATP-gated P2X(4) ion channel in the closed state. Nature, 460, 592–598 (2009).
- 46) Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacol. Rev., 50, 413–492 (1998).
- 47) Khakh BS, Burnstock G, Kennedy C, King BF, North RA, Seguela P, Voigt M, Humphrey PP. International union of pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol. Rev., 53, 107–118 (2001).
- 48) Ulmann L, Hatcher JP, Hughes JP, Chaumont S, Green PJ, Conquet F, Buell GN, Reeve AJ, Chessell IP, Rassendren F. Up-regulation of P2X4 receptors in spinal microglia after peripheral nerve injury mediates BDNF release and neuropathic pain. J. Neurosci., 28, 11263–11268 (2008).
- 49) Tsuda M, Kuboyama K, Inoue T, Nagata K, Tozaki-Saitoh H, Inoue K. Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol. Pain, 5, 1744-8069-5-28 (2009).
- 50) Tsuda M, Inoue K, Salter MW. Neuropathic pain and spinal microglia: a big problem from molecules in “small” glia. Trends Neurosci., 28, 101–107 (2005).
- 51) Österberg A, Boivie J, Thuomas KA. Central pain in multiple sclerosis–prevalence and clinical characteristics. Eur. J. Pain, 9, 531–542 (2005).
- 52) Olechowski CJ, Truong JJ, Kerr BJ. Neuropathic pain behaviours in a chronic-relapsing model of experimental autoimmune encephalomyelitis (EAE). Pain, 141, 156–164 (2009).
- 53) Guo LH, Schluesener HJ. Lesional accumulation of P2X(4) receptor(+) macrophages in rat CNS during experimental autoimmune encephalomyelitis. Neuroscience, 134, 199–205 (2005).
- 54) Vázquez-Villoldo N, Domercq M, Martin A, Llop J, Gomez-Vallejo V, Matute C. P2X4 receptors control the fate and survival of activated microglia. Glia, 62, 171–184 (2014).
- 55) Zhang Z, Zhang ZY, Fauser U, Schluesener HJ. Mechanical allodynia and spinal up-regulation of P2X4 receptor in experimental autoimmune neuritis rats. Neuroscience, 152, 495–501 (2008).
- 56) Matsumura Y, Yamashita T, Sasaki A, Nakata E, Kohno K, Masuda T, Tozaki-Saitoh H, Imai T, Kuraishi Y, Tsuda M, Inoue K. A novel P2X4 receptor-selective antagonist produces anti-allodynic effect in a mouse model of herpetic pain. Sci. Rep., 6, 32461 (2016).
- 57) Sorge RE, Mapplebeck JC, Rosen S, et al. Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat. Neurosci., 18, 1081–1083 (2015).
- 58) Sorge RE, LaCroix-Fralish ML, Tuttle AH, Sotocinal SG, Austin JS, Ritchie J, Chanda ML, Graham AC, Topham L, Beggs S, Salter MW, Mogil JS. Spinal cord Toll-like receptor 4 mediates inflammatory and neuropathic hypersensitivity in male but not female mice. J. Neurosci., 31, 15450–15454 (2011).
- 59) Taves S, Berta T, Liu DL, Gan S, Chen G, Kim YH, Van de Ven T, Laufer S, Ji RR. Spinal inhibition of p38 MAP kinase reduces inflammatory and neuropathic pain in male but not female mice: Sex-dependent microglial signaling in the spinal cord. Brain Behav. Immun., 55, 70–81 (2016).
- 60) Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc. Natl. Acad. Sci. U.S.A., 102, 5856–5861 (2005).
- 61) Tsuda M, Mizokoshi A, Shigemoto-Mogami Y, Koizumi S, Inoue K. Activation of p38 mitogen-activated protein kinase in spinal hyperactive microglia contributes to pain hypersensitivity following peripheral nerve injury. Glia, 45, 89–95 (2004).
- 62) Gu N, Eyo UB, Murugan M, Peng J, Matta S, Dong H, Wu LJ. Microglial P2Y12 receptors regulate microglial activation and surveillance during neuropathic pain. Brain Behav. Immun., 55, 82–92 (2016).
- 63) Staniland AA, Clark AK, Wodarski R, Sasso O, Maione F, D’Acquisto F, Malcangio M. Reduced inflammatory and neuropathic pain and decreased spinal microglial response in fractalkine receptor (CX3CR1) knockout mice. J. Neurochem., 114, 1143–1157 (2010).
- 64) Jin XH, Wang LN, Zuo JL, Yang JP, Liu SL. P2X4 receptor in the dorsal horn partially contributes to brain-derived neurotrophic factor oversecretion and toll-like receptor-4 receptor activation associated with bone cancer pain. J. Neurosci. Res., 92, 1690–1702 (2014).
- 65) Tamura T, Yanai H, Savitsky D, Taniguchi T. The IRF family transcription factors in immunity and oncogenesis. Annu. Rev. Immunol., 26, 535–584 (2008).
- 66) Masuda T, Tsuda M, Yoshinaga R, Tozaki-Saitoh H, Ozato K, Tamura T, Inoue K. IRF8 is a critical transcription factor for transforming microglia into a reactive phenotype. Cell Reports, 1, 334–340 (2012).
- 67) Masuda T, Iwamoto S, Yoshinaga R, Tozaki-Saitoh H, Nishiyama A, Mak TW, Tamura T, Tsuda M, Inoue K. Transcription factor IRF5 drives P2X4R + -reactive microglia gating neuropathic pain. Nat. Commun., 5, 3771 (2014).
- 68) Masuda T, Iwamoto S, Mikuriya S, Tozaki-Saitoh H, Tamura T, Tsuda M, Inoue K. Transcription factor IRF1 is responsible for IRF8-mediated IL-1beta expression in reactive microglia. J. Pharmacol. Sci., 128, 216–220 (2015).
- 69) Tozaki-Saitoh H, Masuda J, Kawada R, Kojima C, Yoneda S, Masuda T, Inoue K, Tsuda M. Transcription factor MafB contributes to the activation of spinal microglia underlying neuropathic pain development. Glia, 67, 729–740 (2019).
- 70) Biber K, Tsuda M, Tozaki-Saitoh H, Tsukamoto K, Toyomitsu E, Masuda T, Boddeke H, Inoue K. Neuronal CCL21 up-regulates microglia P2X4 expression and initiates neuropathic pain development. EMBO J., 30, 1864–1873 (2011).
- 71) Tsuda M, Masuda T, Kitano J, Shimoyama H, Tozaki-Saitoh H, Inoue K. IFN-gamma receptor signaling mediates spinal microglia activation driving neuropathic pain. Proc. Natl. Acad. Sci. U.S.A., 106, 8032–8037 (2009).
- 72) Tsuda M, Toyomitsu E, Komatsu T, Masuda T, Kunifusa E, Nasu-Tada K, Koizumi S, Yamamoto K, Ando J, Inoue K. Fibronectin/integrin system is involved in P2X(4) receptor upregulation in the spinal cord and neuropathic pain after nerve injury. Glia, 56, 579–585 (2008).
- 73) Tsuda M, Tozaki-Saitoh H, Masuda T, Toyomitsu E, Tezuka T, Yamamoto T, Inoue K. Lyn tyrosine kinase is required for P2X(4) receptor upregulation and neuropathic pain after peripheral nerve injury. Glia, 56, 50–58 (2008).
- 74) Tsuda M, Masuda T, Tozaki-Saitoh H, Inoue K. P2X4 receptors and neuropathic pain. Front. Cell. Neurosci., 7, 191 (2013).
- 75) Nakatsuka T, Gu JG. ATP P2X receptor-mediated enhancement of glutamate release and evoked EPSCs in dorsal horn neurons of the rat spinal cord. J. Neurosci., 21, 6522–6531 (2001).
- 76) Jo YH, Schlichter R. Synaptic corelease of ATP and GABA in cultured spinal neurons. Nat. Neurosci., 2, 241–245 (1999).
- 77) Fam SR, Gallagher CJ, Salter MW. P2Y(1) purinoceptor-mediated Ca(2+) signaling and Ca(2+) wave propagation in dorsal spinal cord astrocytes. J. Neurosci., 20, 2800–2808 (2000).
- 78) Imura Y, Morizawa Y, Komatsu R, Shibata K, Shinozaki Y, Kasai H, Moriishi K, Moriyama Y, Koizumi S. Microglia release ATP by exocytosis. Glia, 61, 1320–1330 (2013).
- 79) Burma NE, Bonin RP, Leduc-Pessah H, Baimel C, Cairncross ZF, Mousseau M, Shankara JV, Stemkowski PL, Baimoukhametova D, Bains JS, Antle MC, Zamponi GW, Cahill CM, Borgland SL, De Koninck Y, Trang T. Blocking microglial pannexin-1 channels alleviates morphine withdrawal in rodents. Nat. Med., 23, 355–360 (2017).
- 80) Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, Yamamoto A, Moriyama Y. Identification of a vesicular nucleotide transporter. Proc. Natl. Acad. Sci. U.S.A., 105, 5683–5686 (2008).
- 81) Masuda T, Ozono Y, Mikuriya S, Kohro Y, Tozaki-Saitoh H, Iwatsuki K, Uneyama H, Ichikawa R, Salter MW, Tsuda M, Inoue K. Dorsal horn neurons release extracellular ATP in a VNUT-dependent manner that underlies neuropathic pain. Nat. Commun., 7, 12529 (2016).
- 82) Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW, De Koninck Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature, 438, 1017–1021 (2005).
- 83) Trang T, Beggs S, Wan X, Salter MW. P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J. Neurosci., 29, 3518–3528 (2009).
- 84) Hildebrand ME, Xu J, Dedek A, Li Y, Sengar AS, Beggs S, Lombroso PJ, Salter MW. Potentiation of synaptic GluN2B NMDAR currents by fyn kinase is gated through BDNF-mediated disinhibition in spinal pain processing. Cell Reports, 17, 2753–2765 (2016).
- 85) Chessell IP, Hatcher JP, Bountra C, Michel AD, Hughes JP, Green P, Egerton J, Murfin M, Richardson J, Peck WL, Grahames CB, Casula MA, Yiangou Y, Birch R, Anand P, Buell GN. Disruption of the P2X7 purinoceptor gene abolishes chronic inflammatory and neuropathic pain. Pain, 114, 386–396 (2005).
- 86) Kobayashi K, Takahashi E, Miyagawa Y, Yamanaka H, Noguchi K. Induction of the P2X7 receptor in spinal microglia in a neuropathic pain model. Neurosci. Lett., 504, 57–61 (2011).
- 87) Kim D, Kim MA, Cho IH, Kim MS, Lee S, Jo EK, Choi SY, Park K, Kim JS, Akira S, Na HS, Oh SB, Lee SJ. A critical role of toll-like receptor 2 in nerve injury-induced spinal cord glial cell activation and pain hypersensitivity. J. Biol. Chem., 282, 14975–14983 (2007).
- 88) Clark AK, Staniland AA, Marchand F, Kaan TK, McMahon SB, Malcangio M. P2X7-dependent release of interleukin-1beta and nociception in the spinal cord following lipopolysaccharide. J. Neurosci., 30, 573–582 (2010).
- 89) Heneka MT, Kummer MP, Latz E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol., 14, 463–477 (2014).
- 90) Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M. Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J. Neurosci., 23, 8692–8700 (2003).
- 91) Reeve AJ, Patel S, Fox A, Walker K, Urban L. Intrathecally administered endotoxin or cytokines produce allodynia, hyperalgesia and changes in spinal cord neuronal responses to nociceptive stimuli in the rat. Eur. J. Pain, 4, 247–257 (2000).
- 92) Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine mechanisms of central sensitization: distinct and overlapping role of interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in regulating synaptic and neuronal activity in the superficial spinal cord. J. Neurosci., 28, 5189–5194 (2008).
- 93) Clark AK, Gruber-Schoffnegger D, Drdla-Schutting R, Gerhold KJ, Malcangio M, Sandkuhler J. Selective activation of microglia facilitates synaptic strength. J. Neurosci., 35, 4552–4570 (2015).
- 94) Miyoshi K, Obata K, Kondo T, Okamura H, Noguchi K. Interleukin-18-mediated microglia/astrocyte interaction in the spinal cord enhances neuropathic pain processing after nerve injury. J. Neurosci., 28, 12775–12787 (2008).
- 95) Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J. Neurosci., 26, 3551–3560 (2006).
- 96) Tsuda M, Kohro Y, Yano T, Tsujikawa T, Kitano J, Tozaki-Saitoh H, Koyanagi S, Ohdo S, Ji RR, Salter MW, Inoue K. JAK-STAT3 pathway regulates spinal astrocyte proliferation and neuropathic pain maintenance in rats. Brain, 134, 1127–1139 (2011).
- 97) Kohro Y, Sakaguchi E, Tashima R, Tozaki-Saitoh H, Okano H, Inoue K, Tsuda M. A new minimally-invasive method for microinjection into the mouse spinal dorsal horn. Sci. Rep., 5, 14306 (2015).
- 98) Kanda H, Kobayashi K, Yamanaka H, Okubo M, Noguchi K. Microglial TNFalpha induces COX2 and PGI2 synthase expression in spinal endothelial cells during neuropathic pain. ENeuro, 4, ENEURO.0064-17.2017 (2017).
- 99) Kronschläger MT, Drdla-Schutting R, Gassner M, Honsek SD, Teuchmann HL, Sandkuhler J. Gliogenic LTP spreads widely in nociceptive pathways. Science, 354, 1144–1148 (2016).
- 100) Liu Y, Zhou LJ, Wang J, Li D, Ren WJ, Peng J, Wei X, Xu T, Xin WJ, Pang RP, Li YY, Qin ZH, Murugan M, Mattson MP, Wu LJ, Liu XG. TNF-alpha differentially regulates synaptic plasticity in the hippocampus and spinal cord by microglia-dependent mechanisms after peripheral nerve injury. J. Neurosci., 37, 871–881 (2017).
- 101) Clark AK, Yip PK, Grist J, Gentry C, Staniland AA, Marchand F, Dehvari M, Wotherspoon G, Winter J, Ullah J, Bevan S, Malcangio M. Inhibition of spinal microglial cathepsin S for the reversal of neuropathic pain. Proc. Natl. Acad. Sci. U.S.A., 104, 10655–10660 (2007).
- 102) Clark AK, Malcangio M. Fractalkine/CX3CR1 signaling during neuropathic pain. Front. Cell. Neurosci., 8, 121 (2014).
- 103) Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav. Immun., 21, 642–651 (2007).
- 104) Jacobson KA, Muller CE. Medicinal chemistry of adenosine, P2Y and P2X receptors. Neuropharmacology, 104, 31–49 (2016).
- 105) Donnelly-Roberts D, McGaraughty S, Shieh CC, Honore P, Jarvis MF. Painful purinergic receptors. J. Pharmacol. Exp. Ther., 324, 409–415 (2008).
- 106) Balázs B, Dankó T, Kovács G, Köles L, Hediger MA, Zsembery A. Investigation of the inhibitory effects of the benzodiazepine derivative, 5-BDBD on P2X4 purinergic receptors by two complementary methods. Cell. Physiol. Biochem., 32, 11–24 (2013).
- 107) Hernandez-Olmos V, Abdelrahman A, El-Tayeb A, Freudendahl D, Weinhausen S, Muller CE. N-Substituted phenoxazine and acridone derivatives: structure–activity relationships of potent P2X4 receptor antagonists. J. Med. Chem., 55, 9576–9588 (2012).
- 108) Ase AR, Honson NS, Zaghdane H, Pfeifer TA, Seguela P. Identification and characterization of a selective allosteric antagonist of human P2X4 receptor channels. Mol. Pharmacol., 87, 606–616 (2015).
- 109) Igawa T, Higashi S, Abe Y, Ohkuri T, Tanaka H, Morimoto S, Yamashita T, Tsuda M, Inoue K, Ueda T. Preparation and characterization of a monoclonal antibody against the refolded and functional extracellular domain of rat P2X4 receptor. J. Biochem., 153, 275–282 (2013).
- 110) Igawa T, Kishikawa S, Abe Y, Yamashita T, Nagai S, Shiroishi M, Shinozaki C, Tanaka H, Tozaki-Saitoh H, Tsuda M, Inoue K, Ueda T. Evidence for detection of rat P2X4 receptor expressed on cells by generating monoclonal antibodies recognizing the native structure. Purinergic Signal., 15, 27–35 (2019).
- 111) Williams WA, Linley JE, Jones CA, et al. Antibodies binding the head domain of P2X4 inhibit channel function and reverse neuropathic pain. Pain, 160, 1989–2003 (2019).
- 112) Yamashita T, Yamamoto S, Zhang J, Kometani M, Tomiyama D, Kohno K, Tozaki-Saitoh H, Inoue K, Tsuda M. Duloxetine inhibits microglial P2X4 receptor function and alleviates neuropathic pain after peripheral nerve injury. PLOS ONE, 11, e0165189 (2016).
- 113) Kato Y, Hiasa M, Ichikawa R, Hasuzawa N, Kadowaki A, Iwatsuki K, Shima K, Endo Y, Kitahara Y, Inoue T, Nomura M, Omote H, Moriyama Y, Miyaji T. Identification of a vesicular ATP release inhibitor for the treatment of neuropathic and inflammatory pain. Proc. Natl. Acad. Sci. U.S.A., 114, E6297–E6305 (2017).
- 114) Honore P, Donnelly-Roberts D, Namovic MT, Hsieh G, Zhu CZ, Mikusa JP, Hernandez G, Zhong C, Gauvin DM, Chandran P, Harris R, Medrano AP, Carroll W, Marsh K, Sullivan JP, Faltynek CR, Jarvis MF. A-740003 [N-(1-{[(cyanoimino)(5-quinolinylamino)methyl]amino}-2,2-dimethylpropyl)-2-(3,4-dimethoxyphenyl)acetamide], a novel and selective P2X7 receptor antagonist, dose-dependently reduces neuropathic pain in the rat. J. Pharmacol. Exp. Ther., 319, 1376–1385 (2006).
- 115) Bhattacharya A, Wang Q, Ao H, Shoblock JR, Lord B, Aluisio L, Fraser I, Nepomuceno D, Neff RA, Welty N, Lovenberg TW, Bonaventure P, Wickenden AD, Letavic MA. Pharmacological characterization of a novel centrally permeable P2X7 receptor antagonist: JNJ-47965567. Br. J. Pharmacol., 170, 624–640 (2013).
- 116) Lord B, Aluisio L, Shoblock JR, Neff RA, Varlinskaya EI, Ceusters M, Lovenberg TW, Carruthers N, Bonaventure P, Letavic MA, Deak T, Drinkenburg W, Bhattacharya A. Pharmacology of a novel central nervous system-penetrant P2X7 antagonist JNJ-42253432. J. Pharmacol. Exp. Ther., 351, 628–641 (2014).
- 117) Nieto FR, Clark AK, Grist J, Hathway GJ, Chapman V, Malcangio M. Neuron-immune mechanisms contribute to pain in early stages of arthritis. J. Neuroinflammation, 13, 96 (2016).
- 118) Bhattacharya A, Biber K. The microglial ATP-gated ion channel P2X7 as a CNS drug target. Glia, 64, 1772–1787 (2016).
- 119) Hewitt E, Pitcher T, Rizoska B, Tunblad K, Henderson I, Sahlberg BL, Grabowska U, Classon B, Edenius C, Malcangio M, Lindstrom E. Selective cathepsin S inhibition with MIV-247 Attenuates mechanical allodynia and enhances the antiallodynic effects of gabapentin and pregabalin in a mouse model of neuropathic pain. J. Pharmacol. Exp. Ther., 358, 387–396 (2016).
- 120) Griffin RS, Costigan M, Brenner GJ, Ma CH, Scholz J, Moss A, Allchorne AJ, Stahl GL, Woolf CJ. Complement induction in spinal cord microglia results in anaphylatoxin C5a-mediated pain hypersensitivity. J. Neurosci., 27, 8699–8708 (2007).
- 121) Moriconi A, Cunha TM, Souza GR, et al. Targeting the minor pocket of C5aR for the rational design of an oral allosteric inhibitor for inflammatory and neuropathic pain relief. Proc. Natl. Acad. Sci. U.S.A., 111, 16937–16942 (2014).
- 122) Ohgidani M, Kato TA, Setoyama D, Sagata N, Hashimoto R, Shigenobu K, Yoshida T, Hayakawa K, Shimokawa N, Miura D, Utsumi H, Kanba S. Direct induction of ramified microglia-like cells from human monocytes: dynamic microglial dysfunction in Nasu-Hakola disease. Sci. Rep., 4, 4957 (2014).
- 123) Sellgren CM, Sheridan SD, Gracias J, Xuan D, Fu T, Perlis RH. Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors. Mol. Psychiatry, 22, 170–177 (2017).
- 124) Ohgidani M, Kato TA, Hosoi M, Tsuda M, Hayakawa K, Hayaki C, Iwaki R, Sagata N, Hashimoto R, Inoue K, Sudo N, Kanba S. Fibromyalgia and microglial TNF-alpha: translational research using human blood induced microglia-like cells. Sci. Rep., 7, 11882 (2017).
- 125) Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol. Rev., 63, 772–810 (2011).
- 126) Leduc-Pessah H, Weilinger NL, Fan CY, Burma NE, Thompson RJ, Trang T. Site-specific regulation of P2X7 receptor function in microglia gates morphine analgesic tolerance. J. Neurosci., 37, 10154–10172 (2017).
- 127) Wang Z, Ma W, Chabot JG, Quirion R. Cell-type specific activation of p38 and ERK mediates calcitonin gene-related peptide involvement in tolerance to morphine-induced analgesia. FASEB J., 23, 2576–2586 (2009).
- 128) Horvath RJ, Romero-Sandoval EA, De Leo JA. Inhibition of microglial P2X4 receptors attenuates morphine tolerance, Iba1, GFAP and mu opioid receptor protein expression while enhancing perivascular microglial ED2. Pain, 150, 401–413 (2010).
- 129) Zhou D, Chen ML, Zhang YQ, Zhao ZQ. Involvement of spinal microglial P2X7 receptor in generation of tolerance to morphine analgesia in rats. J. Neurosci., 30, 8042–8047 (2010).
- 130) Ferrini F, Trang T, Mattioli TA, Laffray S, Del’guidice T, Lorenzo LE, Castonguay A, Doyon N, Zhang W, Godin AG, Mohr D, Beggs S, Vandal K, Beaulieu JM, Cahill CM, Salter MW, De Koninck Y. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl(-) homeostasis. Nat. Neurosci., 16, 183–192 (2013).
- 131) Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR, Sotoudeh C, Clark JD, Barres BA, Bohlen CJ, Scherrer G. Loss of mu opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med., 23, 164–173 (2017).
- 132) Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Sagar, Scheiwe C, Nessler S, Kunz P, van Loo G, Coenen VA, Reinacher PC, Michel A, Sure U, Gold R, Grün D, Priller J, Stadelmann C, Prinz M. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature, 566, 388–392 (2019).