Regular Articles
Mouse Pharmacokinetics and in Vitro Metabolism of (±)-Cremastranone
2019 Volume 42 Issue 2 Pages 187-193
Details
2019 Volume 42 Issue 2 Pages 187-193
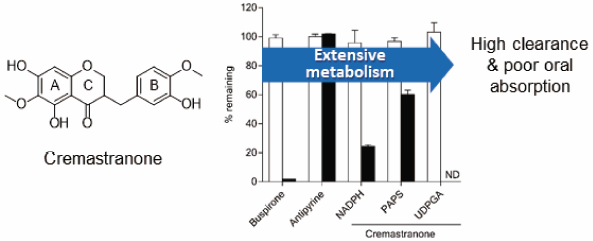
The objective of this study was to characterize pharmacokinetics and metabolism of (±)-cremastranone (CMT) in mouse. Plasma concentrations of CMT following a single oral dose (10 mg/kg) were all below quantitation limit throughout 24-h time course, indicating poor oral bioavailability. Its plasma levels declined rapidly, with a half-life (t1/2) of 1.5 ± 0.3 min following a single intravenous dose (5 mg/kg). They were below the quantitation limit after 15 min post-dosing. CMT showed a high plasma clearance (CLp) of 7.73 ± 3.09 L/h/kg. Consistently, CMT was metabolized rapidly, with a t1/2 < 1 min when it was incubated with liver or intestine S9 fractions of mouse and human in the presence of cofactors for CYP450, uridine 5′-diphosphate (UDP)-glucuronosyltransferase (UGT), and sulfotransferase (ST). Further studies showed that CMT was metabolized by CYP450, UGT, and ST in vitro in liver S9 fractions of mouse and human, with UGT being the major enzyme responsible for its rapid metabolism. CMT was metabolized by UGT and ST in intestine S9 fractions of mouse and human. Mono-demethylated (M1), mono-glucuronide (M2), and mono-sulfate (M3 and M4) metabolites were tentatively identified in vitro. In conclusion, the pharmacokinetics of CMT is suboptimal as a systemic agent, especially as an oral therapy, due to its extensive metabolism. This report provides possible structural modifications to design CMT derivatives with better pharmacokinetic properties.