Abstract
Orthotopic liver transplantation, rather than drug therapy, is the major curative approach for various inherited metabolic disorders of the liver. However, the scarcity of donated livers is a serious problem. To resolve this, there is an urgent need for novel drugs to treat inherited metabolic disorders of the liver. This requirement, in turn, necessitates the establishment of suitable disease models for many inherited metabolic disorders of the liver that currently lack such models for drug development. Recent studies have shown that human induced pluripotent stem (iPS) cells generated from patients with inherited metabolic disorders of the liver are an ideal cell source for models that faithfully recapitulate the pathophysiology of inherited metabolic disorders of the liver. By using patient iPS cell-derived hepatocyte-like cells, drug efficacy evaluation and drug screening can be performed. In addition, genome editing technology has enabled us to generate functionally recovered patient iPS cell-derived hepatocyte-like cells in vitro. It is also possible to identify the genetic mutations responsible for undiagnosed liver diseases using iPS cell and genome editing technologies. Finally, a combination of exhaustive analysis, iPS cells, and genome editing technologies would be a powerful approach to accelerate the identification of novel genetic mutations responsible for undiagnosed liver diseases. In this review, we will discuss the usefulness of iPS cell and genome editing technologies in the field of inherited metabolic disorders of the liver, such as alpha-1 antitrypsin deficiency and familial hypercholesterolemia.
1. INTRODUCTION
The liver plays central roles in maintaining metabolic homeostasis. Therefore, inherited metabolic disorders of the liver (IMDs) can be life-threatening. Many IMD patients require liver transplantation for treatment of their severe metabolic disorders, but not all patients indicated for transplantation receive it due to a shortage of donated livers. As an alternative to liver transplantation, a drug or drugs effective for the treatment of IMDs are highly anticipated. To facilitate the pharmaceutical research, a disease model that faithfully recapitulates the pathophysiology of IMD patients will be indispensable. For many IMDs, however, no such disease model exists, and it can be difficult to obtain biological samples from IMD patients, especially those with rare IMDs. Human induced pluripotent stem (iPS) cell technology is expected to solve these problems.1) Human iPS cells can be easily established from somatic cells such as fibroblasts of IMD patients and proliferate indefinitely. Accordingly, by establishing IMD patient-derived iPS cells (IMD-iPS cells), it would be possible to obtain abundant patient samples for pharmaceutical research. Many groups including ours have developed an efficient hepatocyte differentiation protocol by adopting genetic manipulation,2–6) three-dimensional culture,7–9) co-culture,10–12) extra-cellular matrix,13–15) or small-molecule compounds.16–18) Thus, it is possible to generate human iPS cell-derived hepatocyte-like cells (HLCs) which recapitulate the hepatic functionality in vivo.19–22) Therefore, it is suggested that HLCs derived from IMD-iPS cells would be a useful cell model in terms of drug development.
Among various IMDs, alpha-1 antitrypsin (AAT) deficiency and familial hypercholesterolemia (FH) have been the most actively studied. AAT deficiency (AATD) is a disease caused by mutations in the AAT gene. AATD can cause severe liver diseases, including liver cirrhosis and hepatocellular carcinoma.23) The incidence of AATD is 1 in 1800 to 2000 live births. The PiZ variant (Z-AAT) is the most common of the medically significant null variants, and it is caused by a (G > A) point mutation at codon 342 (Glu342Lys) in exon 5 of the AAT gene.24) The mutation promotes spontaneous Z-AAT polymerization and retention of the Z-AAT polymers in the endoplasmic reticulum (ER) of hepatocytes, resulting in protein overload that causes the liver diseases.25) FH is an autosomal dominant hypercholesterolemia caused by mutations in the low-density lipoprotein receptor (LDLR) gene or LDLR-related genes. FH is characterized by an elevation of serum low density lipoprotein (LDL)-cholesterol which lead to xanthoma formation and premature cardiovascular disease.26,27) The incidence of compound heterozygous or homozygous mutations in the LDLR gene or LDLR-related genes (referred to collectively as HoFH) is 1 in 1500000 to 300000.28) The phenotype of HoFH is much severer than those of heterozygous FH (HeFH). It is also reported that statins are ineffective in HoFH, despite their effectiveness in HeFH.28) In this report, we will overview recent progress in the pharmaceutical research for IMDs, such as AATD and FH, using human iPS cell and genome editing technologies.
2. PHARMACEUTICAL RESEARCH USING PATIENT IPS CELLS
In 2010, to the best of our knowledge, two groups were the first to succeed in establishing human iPS cells from patients with IMDs, including Crigler–Najjar Syndrome, AATD, and FH.29,30) Thereafter, human iPS cells were established from various patients with IMDs (Table 1), and the IMD-iPS cells have contributed to the disease modeling and drug screening for IMDs.
Table 1. Successful Derivations of iPS Cell Lines from Patients with Inherited Metabolic Disorders of the Liver
| Year | Journal | Authors | Disease | Ref. |
|---|
| 2010 | Stem Cell Reviews and Reports | Ghodsizadeh et al. | Tyrosinemia type 1 | 29) |
| Glycogen storage type Ib |
| Progressive familial hereditary cholestasis |
| Crigler–Najjar syndrome |
| 2010 | The Journal of Clinical Investigation | Rashid et al. | Alpha-1 antitrypsin deficiency | 30) |
| Glycogen storage disease type 1a |
| Familial hypercholesterolemia |
| Crigler–Najjar syndrome |
| Hereditary tyrosinemia type 1 |
| 2012 | Hepatology | Cayo et al. | Familial hypercholesterolemia | 31) |
| 2013 | Hepatology | Choi et al. | Alpha-1 antitrypsin deficiency | 32) |
| 2015 | Hepatology | Li et al. | Alpers–Huttenlocher syndrome | 33) |
| 2015 | Hepatology | Tafaleng et al. | Alpha-1 antitrypsin deficiency | 34) |
| 2015 | Stem Cell Reports | Wilson et al. | Alpha-1 antitrypsin deficiency | 35) |
| 2015 | Stem Cells | Soga et al. | Niemann–Pick disease type C | 36) |
| 2017 | EBioMedicine | Bi et al. | Tangier disease | 37) |
| 2017 | Cell Stem Cell | Cayo et al. | Familial hypercholesterolemia | 38) |
| 2017 | Scientific Reports | Imagawa et al. | BSEP-deficiency (progressive familial intrahepatic cholestasis type 2) | 39) |
| 2017 | Biochemical and Biophysical Research Communications | Yoshitoshi-Uebayashi et al. | Citrullinemia type 1 | 40) |
Rashid et al. were the first to establish human iPS cells from a patient with AATD (AATD-iPS cells), and they differentiated these iPS cells into HLCs (AATD-iPS-HLCs).30) The AATD-iPS-HLCs could recapitulate the key pathological features of AATD, namely retention of the Z-AAT polymers in the ER. Choi et al. conducted drug screening using the AATD-iPS-HLCs.32) Among 3131 clinical drugs (including 2800 drugs that have been approved by U.S. Food and Drug Administration (FDA) or have entered phase II clinical trials), 5 drugs could reduce Z-AAT accumulation in the AATD-iPS-HLCs. Carbamazepine, one of the final 5 hit drugs, was consistently shown to decrease Z-AAT accumulation in AATD model mice.41) It is well known that there is a wide variability in the severity of AATD-mediated liver injury. Some AATD patients suffer from severe liver disease (SLD) that necessitates liver transplantation, while others with the same genetic defect suffer from no liver disease (NLD). To elucidate the cause of this variation, Tafaleng et al. established human iPS cells from both groups of AATD patients (SLD-iPS cells and NLD-iPS cells).34) By means of a pulse-chase labeling assay, they then demonstrated a significant difference in the fate of Z-AAT between the two types of cells. That is, the intracellular Z-AAT disappeared more slowly in SLD-iPS-HLCs (half-time 3.6 ± 0.1 h) than in NLD-iPS-HLCs (half-time 2.2 ± 0.3 h). Moreover, transmission electron microscopy examination demonstrated that globular inclusions, large vesicular structures enveloping Z-AAT, were observed in SLD-iPS-HLCs but not in NLD-iPS-HLCs. These results suggest that the degradative response in AATD patients with NLD might be sufficient to prevent Z-AAT accumulation. In addition, this study provided diagnostic criteria for predicting individual differences in AATD-mediated liver injury. Wilson et al. performed microarray analyses to examine the differences between AATD-iPS-HLCs and healthy donor-derived iPS-HLCs.35) They identified the AATD-specific transcriptomic signature, namely, expression of 135 genes diverged from controls. Additionally, they found that carbamazepine treatment increased the protein expression levels of autophagosome components (LC3-II and LC3-I) and decreased intracellular Z-AAT accumulation in the AATD-iPS-HLCs. This result supports the previous finding in an AATD mouse model,41) and suggests that carbamazepine would be an effective drug for AATD.
Rashid et al. were also the first to establish human iPS cells from a patient with FH (FH-iPS cells), which they differentiated into HLCs (FH-iPS-HLCs).30) Here again, the FH-iPS-HLCs could recapitulate the main pathological features of FH, namely deficient LDL receptor–mediated cholesterol uptake. Although their FH patient was diagnosed by symptoms, family history, and LDLR dysfunction assay, the underlying genetic mutation has since been defined. To develop a clearer understanding of the pathophysiology of FH-iPS-HLCs, Cayo et al. established FH-iPS cells from a patient with the JD mutation (FH-JD iPS cells).31) The FH associated with ‘JD’ is a consequence of compound heterozygous mutations in the LDLR gene, which contains a 5 kb deletion of part of exon 13 and all of exons 14 and 15, a truncation mutation, and an A > G substitution for a tyrosine > cysteine substitution at residue 807 in exon 17, which is unable to internalize LDL.42) This compound heterozygous mutations in the LDLR gene causes the loss of LDLR-mediated LDL-C uptake. Cayo et al. have demonstrated that FH-JD-iPS cell-derived HLCs (FH-JD-iPS-HLCs) have no LDL-C uptake capacity and display a marked elevation in the secretion of apolipoprotein B (apoB)-100.38) Consistent with a previous report that statins are ineffective in HoFH patients, such as FH-JD patients,28) LDL uptake capacity in FH-JD-iPS-HLCs was not recovered by lovastatin treatment. This result suggests that the drug efficacy could be predicted using FH-iPS-HLCs. Furthermore, Cayo et al. performed a drug screen to identify novel drug candidates for FH-JD.38) The screen was performed using 2320 small molecules from the Spectrum Collection drug library, which contains 1300 drugs that have reached clinical trials in the U.S.A, Europe, or Japan. They found that several cardiac glycosides, such as digoxin, ouabain, and proscillaridin, could reduce apoB secretion levels via increased proteolytic turnover. They also confirmed that these cardiac glycosides could reduce serum LDL-C in chimeric mice with humanized livers. Therefore, these drugs would be effective in FH-JD patients whose LDL-C levels cannot be decreased using existing drugs.
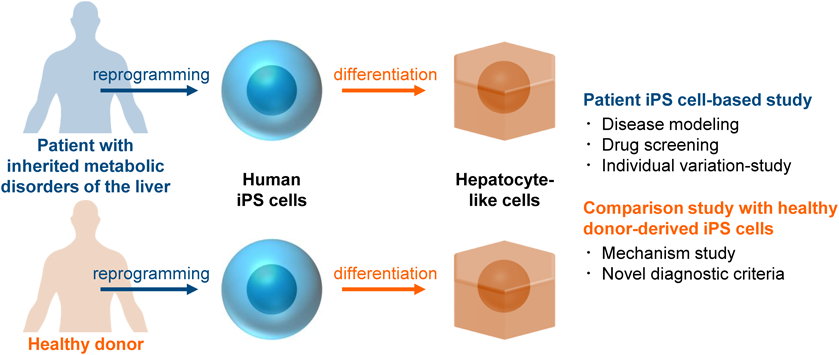
As we mentioned above, patient iPS cells can be a useful disease model for pharmaceutical research (Fig. 1). By conducting a patient iPS cell-based screening assay, new drug candidates might be developed or drug repositioning could be successfully performed. In addition, a personalized medicine could be realized using iPS cells from multiple individuals. Moreover, it is considered that the discovery of novel drug-target molecules will be promoted by performing comprehensive analysis of patient iPS cells.
3. PHARMACEUTICAL RESEARCH USING A COMBINATION OF IPS CELL AND GENOME EDITING TECHNOLOGIES
By introducing genome editing technology into patient iPS cells, the detailed study of inherited genetic disorders has been greatly accelerated. Genome editing technology is based on the use of engineered nucleases including zinc finger nucleases (ZFN),43) transcription activator-like effector nucleases (TALEN),44) and clustered regularly interspaced short palindromic repeats/CRISPR-associated proteins 9 (CRISPR/Cas9) systems.45–48) These chimeric nucleases enable efficient and precise genetic modifications by inducing targeted DNA double-strand breaks that stimulate the cellular DNA repair mechanisms, including nonhomologous end joining (NHEJ) and homology-directed repair (HDR). Until recently, for example, the homologous recombination efficiency in human ES/iPS cells was quite low,49) but it has greatly improved by using these nucleases.50,51) In addition, several genes and chemical compounds with the potential to enhance the genome editing efficiency of human ES/iPS cells have been discovered,52,53) making the genome editing of human ES/iPS cells easier than ever. As a result, many researchers have performed genome editing experiment in IMD-iPS cells (Table 2).
Table 2. Genome Editing Investigations Using iPS Cell Lines from Patients with Inherited Metabolic Disorders of the Liver
| Year | Journal | Authors | Disease | Ref. |
|---|
| 2011 | Nature | Yusa et al. | Alpha-1 antitrypsin deficiency | 54) |
| 2011 | Human Molecular Genetics | Zhang et al. | Wilson’s disease | 55) |
| 2013 | Molecular Biotechnology | Fattahi et al. | Familial hypercholesterolemia | 56) |
| 2013 | Hepatology | Choi et al. | Alpha-1 antitrypsin deficiency | 32) |
| 2014 | Stem Cell Reports | Maetzel et al. | Niemann–Pick disease type C | 57) |
| 2014 | Proceedings of the National Academy of Sciences of the United States of America | Park et al. | Hemophilia A | 58) |
| 2015 | Molecular Therapy | Smith et al. | Alpha-1 antitrypsin deficiency | 59) |
| 2016 | Molecular Therapy–Nucleic Acids | Lee et al. | Hyperargininemia | 60) |
| 2017 | Hepatology Communications | Omer et al. | Familial hypercholesterolemia | 61) |
| 2017 | Cell Reports | Liu et al. | Abetalipoproteinemia | 62) |
| 2018 | Stem Cell Research & Therapy | Lyu et al. | Hemophilia B | 63) |
| 2018 | Cell Reports | Ramaswamy et al. | Hemophilia B | 64) |
| 2018 | Journal of Hepatology | Segeritz et al. | Alpha-1 antitrypsin deficiency | 65) |
To the best of our knowledge, targeted gene correction of AATD-iPS cells was first reported by Yusa et al.54) By using ZFN, they corrected a point mutation (Glu342Lys) in the AAT locus that is responsible for Z-AAT production. Enzyme-linked immunosorbent assay (ELISA) analysis revealed the absence of mutant polymeric Z-AAT and efficient secretion of normal monomeric AAT in the culture supernatant of corrected AATD-iPS-HLCs. Choi et al. later performed a similar gene correction experiment using TALEN instead of ZFN at higher efficiency (the efficiencies were 4 and 25–33% in Yusa et al. and Choi et al., respectively).32) Additionally, CRISPR/Cas9 system-mediated gene correction of AATD-iPS cells has also been reported.59) Recently, Segeritz et al. conducted functional and “omics” comparisons between AATD-iPS-HLCs and genetically corrected AATD-iPS-HLCs to identify new molecular markers and disease signatures.65) They showed that Z-AAT polymer processing was associated with disrupted mitochondrial structure, presence of the oncogenic protein AKR1B10 and two upregulated molecular clusters centered on members of the inflammatory and unfolded protein response pathways. These approaches would be useful for identification of new therapeutic targets for the treatment of AATD.
In addition to correction of AATD-iPS cells, genome editing technology contributes to the functional correction of FH-iPS-HLCs. Omer et al. used a CRISPR/Cas9 system to perform gene correction of iPS cells derived from an HoFH patient (HoFH-iPS cells)61) with a 3-base pair homozygous deletion in exon 4 of the LDLR gene, which results in <5% LDLR activity.66) They adopted a double-nicking strategy using Cas9 nickase (Cas9n). The double-nicking strategy is a novel approach that combines a Cas9n with paired guide RNAs to introduce targeted double-strand breaks.67) This strategy has been proven effective for reducing off-target mutations.68) As expected, sequencing of gene-corrected HoFH-iPS cells compared to non-corrected cells revealed no changes in the 6 selected regions of the genome that had the highest potential for off-targeting.61) Thereafter, they demonstrated that CRISPR-mediated genetic correction could be successfully used to recover the function of cholesterol metabolism in HoFH-iPS cell-derived HLCs. Finally, in 2017, Pashos et al. used iPS cell and genome editing technologies to examine the function of lipid-associated genetic mutations that were discovered by a genome-wide association study (GWAS) and were potentially responsible for FH.69)
Collectively, the above findings clearly demonstrate that functional correction of patient iPS-HLCs can be achieved by using genome editing technology (Fig. 2). In addition, a comparison between gene-corrected cells and non-corrected cells could lead to the discovery of new therapeutic target molecules. Moreover, empirical analysis based on patient iPS cells and genome editing technology would accelerate the identification of disease-related genetic mutations.
4. CONCLUSION
Although we mainly discussed IMDs which show hepatocyte dysfunction, there are also some IMDs caused by dysfunction in liver nonparenchymal cells. It was recently reported that hepatic satellite cells can be generated from human iPS cells.70,71) This technology would be useful for modeling the hepatic fibrosis in IMDs such as congenital hepatic fibrosis (CHF). Several IMDs exert symptoms not only in the liver, but also in other organs. For example, Niemann-Pick disease type C patients suffer from splenomegaly and neurologic disorders in addition to liver disorder. For modeling of such IMDs, characterization of multiple cell types will be necessary.
The disease modeling and drug screening for IMDs can be successfully performed by using patient iPS cells. In addition, functional correction and comparison studies have been progressing steadily by introducing genome editing technology into IMD-iPS cells. We hope that these technologies will reveal new therapeutic drugs for IMDs that cannot be treated using existing drugs.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell, 131, 861–872 (2007).
- 2) Takayama K, Inamura M, Kawabata K, Tashiro K, Katayama K, Sakurai F, Hayakawa T, Furue MK, Mizuguchi H. Efficient and directive generation of two distinct endoderm lineages from human ESCs and iPSCs by differentiation stage-specific SOX17 transduction. PLoS ONE, 6, e21780 (2011).
- 3) Takayama K, Inamura M, Kawabata K, Katayama K, Higuchi M, Tashiro K, Nonaka A, Sakurai F, Hayakawa T, Kusuda Furue M, Mizuguchi H. Efficient generation of functional hepatocytes from human embryonic stem cells and induced pluripotent stem cells by HNF4α transduction. Mol. Ther., 20, 127–137 (2012).
- 4) Takayama K, Inamura M, Kawabata K, Sugawara M, Kikuchi K, Higuchi M, Nagamoto Y, Watanabe H, Tashiro K, Sakurai F, Hayakawa T, Furue MK, Mizuguchi H. Generation of metabolically functioning hepatocytes from human pluripotent stem cells by FOXA2 and HNF1α transduction. J. Hepatol., 57, 628–636 (2012).
- 5) Nakamori D, Takayama K, Nagamoto Y, Mitani S, Sakurai F, Tachibana M, Mizuguchi H. Hepatic maturation of human iPS cell-derived hepatocyte-like cells by ATF5, c/EBPα, and PROX1 transduction. Biochem. Biophys. Res. Commun., 469, 424–429 (2016).
- 6) Mitani S, Takayama K, Nagamoto Y, Imagawa K, Sakurai F, Tachibana M, Sumazaki R, Mizuguchi H. Human ESC/iPSC-derived hepatocyte-like cells achieve zone-specific hepatic properties by modulation of WNT signaling. Mol. Ther., 25, 1420–1433 (2017).
- 7) Takayama K, Kawabata K, Nagamoto Y, Kishimoto K, Tashiro K, Sakurai F, Tachibana M, Kanda K, Hayakawa T, Furue MK, Mizuguchi H. 3D spheroid culture of hESC/hiPSC-derived hepatocyte-like cells for drug toxicity testing. Biomaterials, 34, 1781–1789 (2013).
- 8) Gieseck RL III, Hannan NRF, Bort R, Hanley NA, Drake RAL, Cameron GWW, Wynn TA, Vallier L. Maturation of induced pluripotent stem cell derived hepatocytes by 3D-culture. PLOS ONE, 9, e86372 (2014).
- 9) Yamashita T, Takayama K, Sakurai F, Mizuguchi H. Billion-scale production of hepatocyte-like cells from human induced pluripotent stem cells. Biochem. Biophys. Res. Commun., 496, 1269–1275 (2018).
- 10) Nagamoto Y, Tashiro K, Takayama K, Ohashi K, Kawabata K, Sakurai F, Tachibana M, Hayakawa T, Furue MK, Mizuguchi H. The promotion of hepatic maturation of human pluripotent stem cells in 3D co-culture using type I collagen and Swiss 3T3 cell sheets. Biomaterials, 33, 4526–4534 (2012).
- 11) Berger DR, Ware BR, Davidson MD, Allsup SR, Khetani SR. Enhancing the functional maturity of induced pluripotent stem cell-derived human hepatocytes by controlled presentation of cell–cell interactions in vitro. Hepatology, 61, 1370–1381 (2015).
- 12) Ma X, Qu X, Zhu W, Li Y-S, Yuan S, Zhang H, Liu J, Wang P, Lai CSE, Zanella F, Feng G-S, Sheikh F, Chien S, Chen S. Deterministically patterned biomimetic human iPSC-derived hepatic model via rapid 3D bioprinting. Proc. Natl. Acad. Sci. U.S.A., 113, 2206–2211 (2016).
- 13) Takayama K, Nagamoto Y, Mimura N, Tashiro K, Sakurai F, Tachibana M, Hayakawa T, Kawabata K, Mizuguchi H. Long-term self-renewal of human ES/iPS-derived hepatoblast-like cells on human laminin 111-coated dishes. Stem Cell Reports, 1, 322–335 (2013).
- 14) Cameron K, Tan R, Schmidt-Heck W, Campos G, Lyall MJ, Wang Y, Lucendo-Villarin B, Szkolnicka D, Bates N, Kimber SJ, Hengstler JG, Godoy P, Forbes SJ, Hay DC. Recombinant laminins drive the differentiation and self-organization of hESC-derived hepatocytes. Stem Cell Reports, 5, 1250–1262 (2015).
- 15) Takayama K, Akita N, Mimura N, Akahira R, Taniguchi Y, Ikeda M, Sakurai F, Ohara O, Morio T, Sekiguchi K, Mizuguchi H. Generation of safe and therapeutically effective human induced pluripotent stem cell-derived hepatocyte-like cells for regenerative medicine. Hepatol. Commun., 1, 1058–1069 (2017).
- 16) Ogawa S, Surapisitchat J, Virtanen C, Ogawa M, Niapour M, Sugamori KS, Wang S, Tamblyn L, Guillemette C, Hoffmann E, Zhao B, Strom S, Laposa RR, Tyndale RF, Grant DM, Keller G. Three-dimensional culture and cAMP signaling promote the maturation of human pluripotent stem cell-derived hepatocytes. Development, 140, 3285–3296 (2013).
- 17) Shan J, Schwartz RE, Ross NT, Logan DJ, Thomas D, Duncan SA, North TE, Goessling W, Carpenter AE, Bhatia SN. Identification of small molecules for human hepatocyte expansion and iPS differentiation. Nat. Chem. Biol., 9, 514–520 (2013).
- 18) Nakamae S, Toba Y, Takayama K, Sakurai F, Mizuguchi H. Nanaomycin A treatment promotes hepatoblast differentiation from human iPS cells. Stem Cells Dev., 27, 405–414 (2018).
- 19) Hay DC, Zhao D, Fletcher J, Hewitt ZA, McLean D, Urruticoechea-Uriguen A, Black JR, Elcombe C, Ross JA, Wolf R, Cui W. Efficient differentiation of hepatocytes from human embryonic stem cells exhibiting markers recapitulating liver development in vivo. Stem Cells, 26, 894–902 (2008).
- 20) Si-Tayeb K, Noto FK, Nagaoka M, Li J, Battle MA, Duris C, North PE, Dalton S, Duncan SA. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology, 51, 297–305 (2010).
- 21) Baxter M, Withey S, Harrison S, Segeritz C-P, Zhang F, Atkinson-Dell R, Rowe C, Gerrard DT, Sison-Young R, Jenkins R, Henry J, Berry AA, Mohamet L, Best M, Fenwick SW, Malik H, Kitteringham NR, Goldring CE, Piper Hanley K, Vallier L, Hanley NA. Phenotypic and functional analyses show stem cell-derived hepatocyte-like cells better mimic fetal rather than adult hepatocytes. J. Hepatol., 62, 581–589 (2015).
- 22) Takayama K, Morisaki Y, Kuno S, Nagamoto Y, Harada K, Furukawa N, Ohtaka M, Nishimura K, Imagawa K, Sakurai F, Tachibana M, Sumazaki R, Noguchi E, Nakanishi M, Hirata K, Kawabata K, Mizuguchi H. Prediction of interindividual differences in hepatic functions and drug sensitivity by using human iPS-derived hepatocytes. Proc. Natl. Acad. Sci. U.S.A., 111, 16772–16777 (2014).
- 23) Perlmutter DH. Pathogenesis of chronic liver injury and hepatocellular carcinoma in alpha-1-antitrypsin deficiency. Pediatr. Res., 60, 233–238 (2006).
- 24) Perlmutter DH. Autophagic disposal of the aggregation-prone protein that causes liver inflammation and carcinogenesis in α-1-antitrypsin deficiency. Cell Death Differ., 16, 39–45 (2009).
- 25) Eriksson S, Hagerstrand I. Cirrhosis and malignant hepatoma in alpha 1-antitrypsin deficiency. Acta Med. Scand., 195, 451–458 (1974).
- 26) Rader DJ, Cohen J, Hobbs HH. Monogenic hypercholesterolemia: new insights in pathogenesis and treatment. J. Clin. Invest., 111, 1795–1803 (2003).
- 27) Harada-Shiba M, Arai H, Ishigaki Y, Ishibashi S, Okamura T, Ogura M, Dobashi K, Nohara A, Bujo H, Miyauchi K, Yamashita S, Yokote K, Hypercholesterolemia WG by JAS for MG of F. Guidelines for diagnosis and treatment of familial hypercholesterolemia 2017. J. Atheroscler. Thromb., 25, 751–770 (2018).
- 28) Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen-Thiessen E, Tybjaerg-Hansen A, Watts GF, Averna M, Boileau C, Boren J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AFH, Stroes E, Taskinen M-R, Wiegman A, Wiklund O, Chapman MJ, Cuchel M, Bruckert E, Chapman MJ, Descamps OS, Ginsberg HN, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Raal FJ, Santos RD, Steinhagen-Thiessen E, Tybjaerg-Hansen A, Watts GF, Chapman MJ, Ginsberg HN, Averna M, Boileau C, Boren J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AFH, Stroes E, Taskinen M-R, Wiegman A, Wiklund O, European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur. Heart J., 35, 2146–2157 (2014).
- 29) Ghodsizadeh A, Taei A, Totonchi M, Seifinejad A, Gourabi H, Pournasr B, Aghdami N, Malekzadeh R, Almadani N, Salekdeh GH, Baharvand H. Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells. Stem Cell Rev. Reports, 6, 622–632 (2010).
- 30) Rashid ST, Corbineau S, Hannan N, Marciniak SJ, Miranda E, Alexander G, Huang-doran I, Griffin J, Ahrlund-richter L, Skepper J, Semple R, Weber A, Lomas DA, Vallier L. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Invest., 120, 3127–3136 (2010).
- 31) Cayo MA, Cai J, DeLaForest A, Noto FK, Nagaoka M, Clark BS, Collery RF, Si-Tayeb K, Duncan SA. JD induced pluripotent stem cell-derived hepatocytes faithfully recapitulate the pathophysiology of familial hypercholesterolemia. Hepatology, 56, 2163–2171 (2012).
- 32) Choi SM, Kim Y, Shim JS, Park JT, Wang RH, Leach SD, Liu JO, Deng C, Ye Z, Jang YY. Efficient drug screening and gene correction for treating liver disease using patient-specific stem cells. Hepatology, 57, 2458–2468 (2013).
- 33) Li S, Guo J, Ying Z, Chen S, Yang L, Chen K, Long Q, Qin D, Pei D, Liu X. Valproic acid-induced hepatotoxicity in alpers syndrome is associated with mitochondrial permeability transition pore opening-dependent apoptotic sensitivity in an induced pluripotent stem cell model. Hepatology, 61, 1730–1739 (2015).
- 34) Tafaleng EN, Chakraborty S, Han B, Hale P, Wu W, Soto-Gutierrez A, Feghali-Bostwick CA, Wilson AA, Kotton DN, Nagaya M, Strom SC, Roy-Chowdhury J, Stolz DB, Perlmutter DH, Fox IJ. Induced pluripotent stem cells model personalized variations in liver disease resulting from α1-antitrypsin deficiency. Hepatology, 62, 147–157 (2015).
- 35) Wilson AA, Ying L, Liesa M, Segeritz CP, Mills JA, Shen SS, Jean J, Lonza GC, Liberti DC, Lang AH, Nazaire J, Gower AC, Müeller FJ, Mehta P, Ordóñez A, Lomas DA, Vallier L, Murphy GJ, Mostoslavsky G, Spira A, Shirihai OS, Ramirez MI, Gadue P, Kotton DN. Emergence of a stage-dependent human liver disease signature with directed differentiation of alpha-1 antitrypsin-deficient iPS cells. Stem Cell Reports, 4, 873–885 (2015).
- 36) Soga M, Ishitsuka Y, Hamasaki M, Yoneda K, Furuya H, Matsuo M, Ihn H, Fusaki N, Nakamura K, Nakagata N, Endo F, Irie T, Era T. HPGCD outperforms HPBCD as a potential treatment for Niemann-Pick disease type C during disease modeling with iPS cells. Stem Cells, 33, 1075–1088 (2015).
- 37) Bi X, Pashos EE, Cuchel M, Lyssenko NN, Hernandez M, Picataggi A, McParland J, Yang W, Liu Y, Yan R, Yu C, DerOhannessian SL, Phillips MC, Morrisey EE, Duncan SA, Rader DJ. ATP-binding cassette transporter A1 deficiency in human induced pluripotent stem cell-derived hepatocytes abrogates HDL biogenesis and enhances triglyceride secretion. EBioMedicine, 18, 139–145 (2017).
- 38) Cayo MA, Mallanna SK, Di Furio F, Jing R, Tolliver LB, Bures M, Urick A, Noto FK, Pashos EE, Greseth MD, Czarnecki M, Traktman P, Yang W, Morrisey EE, Grompe M, Rader DJ, Duncan SA. A drug screen using human iPSC-derived hepatocyte-like cells reveals cardiac glycosides as a potential treatment for hypercholesterolemia. Cell Stem Cell, 20, 478–89.e5 (2017).
- 39) Imagawa K, Takayama K, Isoyama S, Tanikawa K, Shinkai M, Harada K, Tachibana M, Sakurai F, Noguchi E, Hirata K, Kage M, Kawabata K, Sumazaki R, Mizuguchi H. Generation of a bile salt export pump deficiency model using patient-specific induced pluripotent stem cell-derived hepatocyte-like cells. Sci. Rep., 7, 41806 (2017).
- 40) Yoshitoshi-Uebayashi EY, Toyoda T, Yasuda K, Kotaka M, Nomoto K, Okita K, Yasuchika K, Okamoto S, Takubo N, Nishikubo T, Soga T, Uemoto S, Osafune K. Modelling urea-cycle disorder citrullinemia type 1 with disease-specific iPSCs. Biochem. Biophys. Res. Commun., 488, 570–571 (2017).
- 41) Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S, Michalopoulos G, Perlmutter DH. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science, 329, 229–232 (2010).
- 42) Davis CG, Lehrman MA, Russell DW, Anderson RGW, Brown MS, Goldstein JL. The J. D. mutation in familial hypercholesterolemia: amino acid substitution in cytoplasmic domain impedes internalization of LDL receptors. Cell, 45, 15–24 (1986).
- 43) Kim Y-G, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. U.S.A., 93, 1156–1160 (1996).
- 44) Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics, 186, 757–761 (2010).
- 45) Ishino Y, Shinagawa H, Makino K, Amemura M, Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol., 169, 5429–5433 (1987).
- 46) Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science, 337, 816–821 (2012).
- 47) Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–823 (2013).
- 48) Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science, 339, 823–826 (2013).
- 49) Zwaka TP, Thomson JA. Homologous recombination in human embryonic stem cells. Nat. Biotechnol., 21, 319–321 (2003).
- 50) Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, Meng X, Miller JC, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol., 27, 851–857 (2009).
- 51) Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, Zeitler B, Cherone JM, Meng X, Hinkley SJ, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R. Genetic engineering of human pluripotent cells using TALE nucleases. Nat. Biotechnol., 29, 731–734 (2011).
- 52) Song J, Yang D, Xu J, Zhu T, Chen YE, Zhang J. RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat. Commun., 7, 10548 (2016).
- 53) Takayama K, Igai K, Hagihara Y, Hashimoto R, Hanawa M, Sakuma T, Tachibana M, Sakurai F, Yamamoto T, Mizuguchi H. Highly efficient biallelic genome editing of human ES/iPS cells using a CRISPR/Cas9 or TALEN system. Nucleic Acids Res., 45, 5198–5207 (2017).
- 54) Yusa K, Rashid ST, Strick-Marchand H, Varela I, Liu P-Q, Paschon DE, Miranda E, Ordóñez A, Hannan NRF, Rouhani FJ, Darche S, Alexander G, Marciniak SJ, Fusaki N, Hasegawa M, Holmes MC, Di Santo JP, Lomas DA, Bradley A, Vallier L. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells. Nature, 478, 391–394 (2011).
- 55) Zhang S, Chen S, Li W, Guo X, Zhao P, Xu J, Chen Y, Pan Q, Liu X, Zychlinski D, Lu H, Tortorella MD, Schambach A, Wang Y, Pei D, Esteban MA. Rescue of ATP7B function in hepatocyte-like cells from Wilson’s disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin. Hum. Mol. Genet., 20, 3176–3187 (2011).
- 56) Fattahi F, Asgari S, Pournasr B, Seifinejad A, Totonchi M, Taei A, Aghdami N, Salekdeh GH, Baharvand H. Disease-corrected hepatocyte-like cells from familial hypercholesterolemia-induced pluripotent stem cells. Mol. Biotechnol., 54, 863–873 (2013).
- 57) Maetzel D, Sarkar S, Wang H, Abi-Mosleh L, Xu P, Cheng AWW, Gao Q, Mitalipova M, Jaenisch R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from niemann-pick type C patient-specific iPS cells. Stem Cell Reports, 2, 866–880 (2014).
- 58) Park C-Y, Kim J, Kweon J, Son JS, Lee JS, Yoo J-E, Cho S-R, Kim J-H, Kim J-S, Kim D-W. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc. Natl. Acad. Sci. U.S.A., 111, 9253–9258 (2014).
- 59) Smith C, Abalde-Atristain L, He C, Brodsky BR, Braunstein EM, Chaudhari P, Jang Y-Y, Cheng L, Ye Z. Efficient and allele-specific genome editing of disease loci in human iPSCs. Mol. Ther., 23, 570–577 (2015).
- 60) Lee PC, Truong B, Vega-Crespo A, Gilmore WB, Hermann K, Angarita SA, Tang JK, Chang KM, Wininger AE, Lam AK, Schoenberg BE, Cederbaum SD, Pyle AD, Byrne JA, Lipshutz GS. Restoring ureagenesis in hepatocytes by CRISPR/Cas9-mediated genomic addition to arginase-deficient induced pluripotent stem cells. Mol. Ther. Nucleic Acids, 5, e394 (2016).
- 61) Omer L, Hudson EA, Zheng S, Hoying JB, Shan Y, Boyd NL. CRISPR correction of a homozygous low-density lipoprotein receptor mutation in familial hypercholesterolemia induced pluripotent stem cells. Hepatol. Commun., 1, 886–898 (2017).
- 62) Liu Y, Conlon DM, Bi X, Slovik KJ, Shi J, Edelstein HI, Millar JS, Javaheri A, Cuchel M, Pashos EE, Iqbal J, Hussain MM, Hegele RA, Yang W, Duncan SA, Rader DJ, Morrisey EE. Lack of MTTP activity in pluripotent stem cell-derived hepatocytes and cardiomyocytes abolishes apoB secretion and increases cell stress. Cell Reports, 19, 1456–1466 (2017).
- 63) Lyu C, Shen J, Wang R, Gu H, Zhang J, Xue F, Liu X, Liu W, Fu R, Zhang L, Li H, Zhang X, Cheng T, Yang R, Zhang L. Targeted genome engineering in human induced pluripotent stem cells from patients with hemophilia B using the CRISPR-Cas9 system. Stem Cell Res. Ther., 9, 92 (2018).
- 64) Ramaswamy S, Tonnu N, Menon T, Lewis BM, Green KT, Wampler D, Monahan PE, Verma IM. Autologous and heterologous cell therapy for hemophilia B toward functional restoration of factor IX. Cell Reports, 23, 1565–1580 (2018).
- 65) Segeritz C-P, Rashid ST, Cardoso de Brito M, Paola MS, Ordonez A, Morell CM, Kaserman JE, Madrigal P, Hannan N, Gatto L, Tan L, Wilson AA, Lilley K, Marciniak SJ, Gooptu B, Lomas DA, Vallier L. hiPSC hepatocyte model demonstrates the role of unfolded protein response and inflammatory networks in α1-antitrypsin deficiency. J. Hepatol., 69, 851–860 (2018).
- 66) Hobbs HH, Russell DW, Brown MS, Goldstein JL. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu. Rev. Genet., 24, 133–170 (1990).
- 67) Ran FA, Hsu PD, Lin C-Y, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell, 154, 1380–1389 (2013).
- 68) Ishida K, Gee P, Hotta A. Minimizing off-target mutagenesis risks caused by programmable nucleases. Int. J. Mol. Sci., 16, 24751–24771 (2015).
- 69) Pashos EE, Park YS, Wang X, Raghavan A, Yang W, Abbey D, Peters DT, Arbelaez J, Hernandez M, Kuperwasser N, Li W, Lian Z, Liu Y, Lv W, Lytle-Gabbin SL, Marchadier DH, Rogov P, Shi J, Slovik KJ, Stylianou IM, Wang L, Yan R, Zhang X, Kathiresan S, Duncan SA, Mikkelsen TS, Morrisey EE, Rader DJ, Brown CD, Musunuru K. Large, diverse population cohorts of hiPSCs and derived hepatocyte-like cells reveal functional genetic variation at blood lipid-associated Loci. Cell Stem Cell, 20, 558–70.e10 (2017).
- 70) Koui Y, Kido T, Ito T, Oyama H, Chen S-W, Katou Y, Shirahige K, Miyajima A. An In vitro human liver model by iPSC-derived parenchymal and non-parenchymal cells. Stem Cell Reports, 9, 490–498 (2017).
- 71) Coll M, Perea L, Boon R, Leite SB, Vallverdú J, Mannaerts I, Smout A, El Taghdouini A, Blaya D, Rodrigo-Torres D, Graupera I, Aguilar-Bravo B, Chesne C, Najimi M, Sokal E, Lozano JJ, van Grunsven LA, Verfaillie CM, Sancho-Bru P. Generation of hepatic stellate cells from human pluripotent stem cells enables in vitro modeling of liver fibrosis. Cell Stem Cell, 23, 101–113.e7 (2018).