Abstract
Cigarette smoke is a well-known strong risk factor for inducing airway hyperreactivity (AHR), but the underlying molecular mechanisms are not fully understood. In the present study, mouse in-vivo and in-vitro models were used to study effects of dimethyl sulfoxide (DMSO)-extracted cigarette smoke particles (DSP) on the airway, and to explore the underlying molecular mechanisms that are involved in DSP-induced AHR. In mouse in-vivo model, DSP (0.75, 1.5 or 3 µL/mL) was administered intranasally daily for 7 d. At the end of this period, lung functions were measured with flexiVent™. The results showed that the mice exhibited AHR in a dose-dependent manner following methacholine inhalation in vivo. In mouse in-vitro organ culture model, exposure of mouse tracheal segments to DSP (0.1 µL/mL) with or without the following pharmacological inhibitors: specific c-Jun-N-terminal kinase (JNK) inhibitor SP600125 (10 µM) or the anti-inflammatory drug dexamethasone (1 µM). DSP-induced bradykinin receptor-mediated airway contraction with increased mRNA and protein expressions for bradykinin B1 and B2 receptors could be significantly reduced by SP600125 or dexamethasone. In conclusion, the present study demonstrates that DSP could induce AHR in vivo and in vitro. In addition to this, the upregulation of bradykinin receptors in airway is most likely one of the underlying molecular mechanisms involved.
INTRODUCTION
Airway hyperreactivity (AHR) is defined as an abnormal increase in responsiveness to various specific and nonspecific stimuli in airway. It is an important feature of asthmatic inflammatory airways and chronic obstructive pulmonary disease (COPD). Environmental risk factors like cigarette smoke exposure are closely associated with the development of AHR, asthma and COPD. Both active and passive cigarette smoke exposures may affect airway nerves,1) epithelium,2) smooth muscle cells,3) and subsequently to participate in the pathogenesis of airway inflammation, AHR, asthma and COPD.4)
Asthma and COPD are different disease entities. However, there are many overlapping features of these two diseases. Especially in older patients it can be difficult to differentiate between asthma and COPD,5) and therefore it is called the “asthma-COPD overlap syndrome.”6) In China, deaths caused by COPD are mainly related to cigarette smoke exposure.7) One of explanations is that cigarette smoke contains high amounts of particulate matter (PM) 2.5 and is the main source of indoor PM 2.5 pollution.8) PM is one of the most harmful components of air pollution. The smaller the particle size, the deeper it can penetrate our respiratory system and the more harmful it is to our health.9) PM 2.5 pollution from cigarette smoke often suspends in indoor air for a long time and is easier to inhale into the lungs and permanently stays in the alveoli.10)
Studies have shown that long-term exposure to cigarette smoke involves airway smooth muscle cell proliferation, and triggers AHR and remodelling11); exposure to inhaled nicotine-containing e-cigarette fluids leads to increased cytokine expression, AHR and the development of COPD, and more importantly, these effects are nicotine-dependent12); smokers with asthma exhibit neutrophilic airway inflammation in addition to eosinophilic inflammation.13) Using a murine passive smoking model and an organ culture model of exposing bronchial segments to dimethyl sulfoxide (DMSO)-extracted cigarette smoke particles (DSP), we have previously demonstrated that cigarette smoke exposure in vivo or exposure to cigarette smoke extracts in vitro promote smooth muscle proliferation and AHR via upregulation of G-protein coupled receptors (GPCR) such as endothelin receptors and 5-hydroxytryptamine (5-HT) 2A receptors.3,14) The GPCR upregulation is mediated by activation of mitogen-activated protein kinases (MAPK) induced by cigarette smoke exposure, suggesting that MAPK-dependent upregulation of GPCR is a key molecular mechanism that leads to AHR, and therefore may provide an option for treatment of AHR in asthma, COPD as well as in other inflammatory airway diseases.4)
Bradykinin is an important inflammatory mediator.15,16) Bradykinin and its receptors are suggested to be involved in the pathogenesis of asthma and COPD.4) Bradykinin acts via its B1 and B2 receptors and has many effects on the airways including induction of bronchoconstriction, bronchodilation, mucus secretion and oedema, stimulation of cholinergic and sensory nerves.17) Both the B1 and B2 receptors appear to be involved in allergen-induced bronchial hyperreactivity in rat.18) Inhalation of bradykinin induces a strong bronchoconstriction in asthmatic patients,19,20) and intravenous administration causes intense bronchoconstriction in guinea pigs.21) Elevated levels of bradykinin are found in bronchoalveolar lavage fluid from asthmatic patients.22) Thus, bradykinin and des-Arg9-bradykinin and its receptors might play a significant role in asthma, COPD and airway inflammation as well.
Previously, we have showed that nicotine, an important component contained in cigarette smoke, increased the expression of bradykinin B1 and B2 receptor-mediated airway contractions, but does not affect airway contractile responses to 5-HT, acetylcholine or endothelin receptor agonists.23) The present study is designed to demonstrate that DSP can induce AHR in vivo, and furthermore to investigate if upregulation of bradykinin B1 and B2 receptors is involved in DSP-induced AHR in vitro. Understanding the underlying molecular mechanisms that are responsible for cigarette smoke-induced AHR might provide insight into the pathogenesis of smoke-induced AHR seen in patients with COPD and asthma, and aid the development of novel treatment options.
MATERIALS AND METHODS
Extraction of DSPAs previously described,24) cigarettes (Marlboro, 0.8 mg nicotine per cigarette) were ‘smoked’ by an aspirator, and the smoke was directed through a cotton wool filter. The retained smoke particles from three cigarettes in the filter were dissolved in 1 mL of DMSO and diluted to a standard nicotine content of 0.1 mg/mL. These stock solutions of DSP were kept at −20°C for all subsequent experiments.
Mouse in-Vivo ModelAdult male BALB/c mice (10–12 weeks of age, 25 ± 2 g body weight at the time of the study) were purchased from the Fourth Military Medical University (Xi’an, China). The BALB/c mice were housed in a vivarium facility under a 12-h light/12-h dark cycle and were maintained under standard conditions (22 ± 1°C, 45% humidity, food and water ad libitum). The BALB/c mice were randomly divided into the following four groups: control group containing 0.3% DMSO), low DSP dose (0.75 µL/mL) group, middle DSP dose (1.5 µL/mL) group, and high DSP dose (3 µL/mL) group. All groups received daily intranasal administration of DSP. On day 7, the lung mechanics were measured using a flexiVent™ small animal ventilator to study DSP-induced AHR. Animal experiments were initiated after obtaining approval from the Ethics Committee of Xi’an Medical University (Xi’an, China).
Mouse in-Vitro ModelThe present study followed previously published protocol by Zhang et al.25) with a slight modification to study the effects of DSP on contractile response to kinins, and to explore the underlying molecular mechanisms involved. Tracheae from male BALB/c mice were dissected, cut into three or four segments and placed individually into wells of a 96-well plate (Ultra-low attachment) with 300 µL of serum-free Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with penicillin (100 U/mL) and streptomycin (100 µg/mL). The segments were incubated for 2 to 4 d at 37°C in humidified 5% CO2 in the absence or presence of DSP (0.05 or 0.1 µL/mL) with or without the following pharmacological inhibitors: anti-inflammatory drug dexamethasone (1 µM) or specific c-Jun-N-terminal kinase (JNK) inhibitor SP600125 (10 µM). The segments were moved into new wells containing fresh medium, fresh medium with vehicle, fresh medium with DSP, or fresh medium with DSP and pharmacological inhibitors daily.
Lung MechanicsA computer-controlled small animal ventilator (flexiVent™, SCIREQ Inc, Montreal, QC, Canada) was used to measure lung function and assess airway responsiveness to methacholine inhalation in anesthetized mice (sodium pentobarbital, 90 mg/kg body weight) on a heating pad (37°C), according to the manufacturer’s protocol.26) The system applies the forced oscillation technique (FOT) to measure respiratory system parameters and assess AHR following cumulative doses of aerosol inhalation methacholine administration. Measurements are fitted into a constant phase model to compute the following respiratory parameters: 1) resistance of respiratory system (Rrs), elastance of respiratory system (Ers) and dynamic compliance of respiratory system (Crs) by the data obtained from the single frequency FOT, and 2) Newtonian resistance (Rn), tissue damping (G) and tissue elastance (H) by the data acquired from the broadband FOT.
In-Vitro PharmacologyThe experiments were performed as previously described.25) Briefly, the organ-cultured mouse tracheal segments were immersed in a temperature-controlled (37°C) myograph bath (Organ Bath Model 700 MO). Following equilibration, a 0.8 mN pre-tension was applied to each segment. Each segment was contracted by applying 60 mM KCl to test the contractile function. After wash out, the concentration–effect curves of the kinin B1 receptor agonist des-Arg9-bradykinin and kinin B2 receptor agonist bradykinin were then generated by cumulative administration. At the end of the experiment, the acetylcholine (ACh) 50 µM was given to reach the maximum contraction.
Western BlottingThe tracheal samples were frozen on ice for 1 h in RIPA buffer containing 0.5 mM protease inhibitor phenylmethylsulfonyl fluoride (PMSF) (Roche, Basel, Switzerland). The protein concentration was then measured. Each sample was subsequently denatured by boiling (95–98°C) for 5 min in Laemmle loading buffer. The protein samples (40 µg) were loaded, separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. After blocking with 5% bovine serum albumin or non-fat dried milk for 1 h at 37°C, the membranes were incubated with the following antibodies overnight at 4°C: anti-kinin B1 antibody (Santa Cruz Technology, Dallas, TX, U.S.A.), anti-kinin B2 antibody (Santa Cruz Technology) and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (Santa Cruz Technology). After washing, the membranes were incubated with horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (IgG) (Abcam, Cambridge, MA, U.S.A.) for 1 h at 37°C. After three 10-min washes, the membranes were developed using the Working Solution (LumiSensor A + B) for 3 min. The protein bands were analysed using Quantity One 4.6.2 (Bio-Rad, Co., Ltd.).
Real-Time PCRThe tracheal smooth muscle was isolated mechanically on ice tray under a microscope, and total RNA extracted from the smooth muscle was reversely transcribed to cDNA followed previously published protocol by Zhang et al.27) Real-time quantitative PCR was performed with the QuantiTectTM SYBR® Green PCR Kit (QIAGEN, Germany) in a GeneAmp 7300 sequence detection system (Perkin-Elmer, MA, U.S.A.). The data were analysed using the threshold cycle (CT) method, and the specificity of the PCR products was evaluated using dissociation curves. The relative amount of mRNA was calculated by normalizing the CT values of the mRNA levels of the target gene to the housekeeping gene GAPDH in the same sample.
The specific primers for the murine kinin B1 and B2 receptors and GAPDH were designed using Prime Express 2.0 software (Applied Biosystems, Foster City, CA, U.S.A.) and synthesized by DNA Technology A/S (Aarhus, Denmark). The primer sequences are shown in Table 1.
Table 1. Sequences of Specific PCR Primers
| Target | Genebank ID | Sequence (5′–3′) |
|---|
| Kinin B1 receptor | NM_007539 | Fwd: CCA TAG CAGAAA TCT ACC TGG CTA AC |
| Rev: GCC AGT TGA AAC GGT TCC |
| Kinin B2 receptor | NM_009747 | Fwd: ATG TTC AAC GTC ACC ACA CAA GTC |
| Rev: TGG ATG GCA TTG AGC CAA C |
| GAPDH | NM_008084 | Fwd: CAT GGC CTT CCG TGT TCC TA |
| Rev: TGC TTC ACC ACC TTC TTG ATG |
GAPDH, Glyceraldehyde 3-phosphate dehydrogenase.
All data are expressed as the mean values ± standard error of the mean (S.E.M.), and ‘n’ represents the number of animals or experiments performed. Data analysis was performed using GraphPad Prism 5. The concentration–effect curves were fit to sigmoidal dose–response curves. Two-way ANOVA with a Bonferroni post-hoc test was performed to compare the concentration–effect curves for the data from in vivo study. One-way ANOVA with Bonferroni post-hoc test or unpaired Student’s t-test was performed for the data from in vitro study to compare the mRNA and protein data. p < 0.05 was defined as statistically significant.
RESULTS
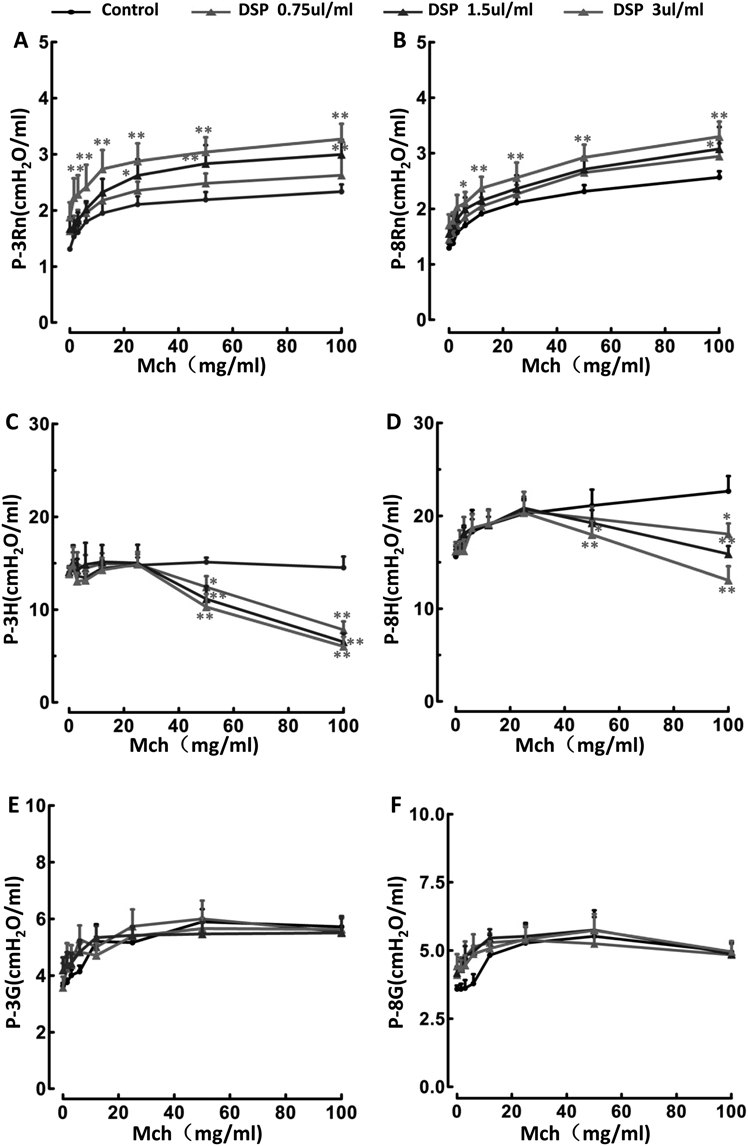
Effects of DSP on Resistance of Respiratory System (Rrs), Elastance of Respiratory System (Ers) and Dynamic Compliance of Respiratory System (Crs) in the in-Vivo ModelThe level of constriction in the lungs following cumulative doses of methacholine inhalation was quantitatively assessed with the data obtained from single frequency FOT on the flexiVent™ system. After intranasal treatment with different concentrations of DSP (0.75, 1.5 or 3 µL/mL) for 7 d, the mice exhibited significantly dose-dependent increases in the Rrs (Fig. 1A, n = 10; p < 0.05, p < 0.01) and Ers (Fig. 1B, n = 10; p < 0.05, p < 0.01), as well as reduction of the Crs (Fig. 1C, n = 10; p < 0.05, p < 0.01), demonstrating that DSP dose-dependently induced AHR in mice in vivo.
However, DSP did not significantly alter quasi-static compliance (Cst, Fig. 2A) or quasi-static elasticity (Est, Fig. 2B) in mice (n = 10, p > 0.05).
Effects of DSP on Newtonian Resistance (Rn), Tissue Damping (G) and Tissue Elastance (H) by the Data Acquired from the Broadband FOTTo further confirm DSP-induced AHR in mice, and differentiate the contribution of the central respective peripheral airways, Rn, G and H were assessed by performing broadband FOT for 3 s (Figs. 3A, C, E) or 8 s (Figs. 3B, D, F) on the flexiVent™. After intranasal administration of DSP for 7 d, there was a significantly concentration-dependent increase in airway resistance in the mice caused by methacholine inhalation in central airways (Newtonian resistance, Rn; Figs. 3A, B; n = 10; p < 0.05), but not in peripheral airways (tissue damping, G, Figs. 3E, F, n = 10, p > 0.05). In addition, DSP significantly reduced tissue elastance (H; Figs. 3C, D; n = 10; p < 0.05).
DSP-Induce Increased Bradykinin (BK) Receptor-Mediated Contractions in a Time- and Concentration-Dependent MannerIn order to assess the time-course of DSP effects on the airway contraction, tracheal segments were organ cultured for 2 or 4 d in the presence of DSP or vehicle (DMSO). The results show that a tendency towards an increased airway contractile response to both des-Arg9-BK (Fig. 4A, Table 2, p > 0.05) and BK (Fig. 4B, Table 2, p > 0.05) was seen after 2 d of DSP (0.1 µL/mL) treatment, compared with control segments that are exposed to the same volume of vehicle (DMSO). After the tracheal segments were treated with DSP for 4 d, the lower DSP concentration (0.05 µL/mL) did not significantly increase contractile responses to des-Arg9-BK (Fig. 4C, Table 2, p > 0.05) and BK (Fig. 4D, Table 2, p > 0.05). But culture with 0.1 µL/mL of DSP significantly increased the Emax and pEC50 for both des-Arg9-BK (Fig. 4C, Table 1, Emax: 17.5 ± 2.0 vs. 35.3 ± 2.4, p < 0.001, pEC50: 7.63 ± 0.32 vs. 8.48 ± 0.18, p < 0.05), and BK (Fig. 4D, Table 2, Emax: 30.6 ± 9.9 vs. 57.1 ± 6.3, p < 0.05, pEC50: 5.44 ± 0.91 vs. 8.36 ± 0.49, p < 0.05), compared with vehicle groups.
Table 2. Effects of DSP on Des-Arg
9-Bradykinin- and Bradykinin-Induced Airway Contractions
| Incubation time | DSP (µL/mL) | Des-Arg9-bradykinin | Bradykinin |
|---|
| n | Emax (% of ACh) | pEC50 | n | Emax (% of ACh) | pEC50 |
|---|
| Day 2 | Vehicle (Ctrl) | 4 | 27.3 ± 3.4 | 7.18 ± 0.27 | 4 | 29.8 ± 7.9 | 5.75 ± 0.88 |
| 0.1 | 4 | 29.7 ± 3.7 | 7.54 ± 0.28 | 4 | 45.9 ± 8.2** | 7.31 ± 0.72* |
| Day 4 | Vehicle (Ctrl) | 8 | 17.5 ± 2.0 | 7.63 ± 0.32 | 7 | 30.6 ± 9.9 | 5.44 ± 0.91 |
| 0.05 | 5 | 20.2 ± 2.7 | 8.02 ± 0.32 | 5 | 50.2 ± 8.3 | 7.44 ± 0.64 |
| 0.1 | 7 | 35.3 ± 2.4** | 8.48 ± 0.18* | 7 | 57.1 ± 6.3* | 8.36 ± 0.49* |
Tracheal segments were cultured for 2 or 4 d in presence of vehicle (0.1% DMSO, Ctrl) or DSP (0.05 or 0.1 µL/mL). Emax and pEC50 for des-Arg9-bradykinin (bradykinin B1 receptor agonist) and bradykinin (bradykinin B2 receptor agonist) are presented as mean ± S.E.M. Statistical analysis was performed using unpaired Student’s t-test. Vehicle (Ctrl) vs. DSP, * p < 0.05, ** p < 0.01, n = number of animals.
The relative amount of mRNA in the smooth muscle for bradykinin B1 and B2 receptors was analyzed by quantitative real-time PCR. Four days of organ culture in the presence of DSP (0.1 µL/mL) significantly enhanced the mRNA expression for both bradykinin B1and B2 receptors, compared to control (Fig. 5, n = 3, p < 0.01).
The corresponding bradykinin B1 and B2 receptor protein expression was examined using Western blotting. Compared to the solvent DMSO, both bradykinin B1 and B2 receptor protein was upregulated in the airways after incubation with DSP for 4 d (Fig. 6, n = 3; p < 0.01).
Effects of SP600125 and Dexamethasone on DSP-Enhanced Bradykinin B1 and B2 Receptor-Mediated Airway Contraction, Receptor mRNA and Protein ExpressionIn order to further elucidate the involvement of the JNK and nuclear factor-kappaB (NF-κB) inflammatory signaling pathway in DSP effects, the effect of the JNK inhibitor SP600125 (10 µM) or anti-inflammatory drug dexamethasone (Dex, 1 µM) on DSP-induced bradykinin B1 and B2 receptor-mediated airway contraction, receptor mRNA and protein expressions were examined. Blockage of JNK signal pathways using SP600125 resulted in a significant reduction of the DSP-enhanced contractile responses to bradykinin B1 receptor-mediated airway contraction (Fig. 7A, Table 3, Emax: 35.3 ± 2.4 vs. 20.8 ± 2.3, p < 0.01; pEC50: 8.48 ± 0.18 vs. 6.79 ± 0.26, p < 0.001). However, SP600125 did not significantly alter the Emax and pEC50 values of bradykinin B2 receptor-mediated airway contraction curves (Fig. 7B, Table 3, Emax: 58.3 ± 7.3 vs. 40.5 ± 2.8, p > 0.05; pEC50: 8.30 ± 0.57 vs. 7.99 ± 0.27, p > 0.05). Anti-inflammatory drug dexamethasone, a general NF-κB pathway inhibitor, almost completely abolished the DSP effects on both brady kinin B1 (Fig. 7A, Table 3) and B2 (Fig. 7B, Table 3) receptor-mediated airway contractions.
Table 3. Effects of SP600125 and Dexamethasone on DSP Enhanced Des-Arg
9-Bradykinin- and Bradykinin-Induced Airway Contractions
| Treatment | Des-Arg9-bradykinin | Bradykinin |
|---|
| n | Emax (% of ACh) | pEC50 | n | Emax (% of ACh) | pEC50 |
|---|
| DSP 0.1 µL/mL + DMSO | 7 | 35.3 ± 2.4 | 8.48 ± 0.18 | 7 | 58.3 ± 7.3 | 8.30 ± 0.57 |
| DSP 0.1 µL/mL + DEX 1 µM | 5 | 13.5 ± 1.2** | 7.12 ± 0.21** | 5 | 28.1 ± 4.8* | 6.10 ± 0.50* |
| DSP 0.1 µL/mL + SP 10 µM | 4 | 20.8 ± 2.3** | 6.79 ± 0.26** | 4 | 40.5 ± 2.8 | 7.99 ± 0.27 |
Tracheal segments were cultured for 4 d in presence of DSP 0.1 µL/mL with SP600125 (SP, 10 µM) or dexamethasone (DEX, 1 µM). Emax and pEC50 for des-Arg9-bradykinin (bradykinin B1 receptor agonist) and bradykinin (bradykinin B2 receptor agonist) are presented as mean ± S.E.M. Statistical analysis was performed using unpaired Student’s t-test. DSP + DMSO vs. DSP + DEX or SP, * p < 0.05, ** p < 0.01, n = number of animals.
In addition, compared to the control, SP600125 or dexamethasone could both significantly inhibit bradykinin B1 and B2 receptor mRNA (Fig. 5) and protein (Fig. 6) expressions in the DSP-treated groups.
DISCUSSION
Airway smooth muscle cells are known to be the main effector cells of airway narrowing in AHR and asthma pathogenesis,28) but knowledge about the underlying molecular mechanisms behind how the smooth muscle cells participate in AHR are limited. The present study, using a mouse in-vivo model has demonstrated that cigarette extract DSP could induce AHR, and using a mouse in-vitro model found that upregulation of bradykinin B1 and B2 receptors were involved in the DSP-induced AHR. In addition, JNK-mediated inflammatory signal pathways were at least partly responsible for the DSP-induced upregulation of bradykinin receptors, and subsequent AHR.
Bradykinin and its related peptides are important inflammatory mediators and play key roles in airway inflammation and AHR.4) There are two major bradykinin receptors: the bradykinin B1 and B2 receptors.4,29) Bradykinin B1 receptor is hardly expressed in the airway of healthy individuals, while bradykinin B2 receptor is constitutively expressed in airway.4,29) In the pathogenic condition, there is induced expression of bradykinin B1 receptor.4,29) Both bradykinin B1 and B2 receptors expressed in airway smooth muscle cells mediate smooth muscle contraction, and airway narrowing as well as AHR.4) Environmental risk factors like cigarette smoke may trigger the production of pro-inflammatory mediators such as tumor necrosis factor-α (TNF-α) and interleukins (ILs) that activate intracellular MAPK- and NF-κB-dependent inflammatory pathways, as well as intracellular JNK-mediated inflammatory pathways, which subsequently lead to upregulation of bradykinin receptors in airway smooth muscle cells and contribute to the development of AHR and airway narrowing.4) In the present study, cigarette smoke extract DSP was administered daily intranasally for 7 d, the mice exhibited AHR in a dose-dependent manner following methacholine inhalation administration in vivo. Specific JNK inhibitor SP600125 or the anti-inflammatory drug dexamethasone significantly inhibited DSP-induced bradykinin receptor-mediated AHR. This demonstrates that DSP could induce AHR and that upregulation of bradykinin receptors is involved in this process, which are well in line with our previous findings that cytokines induce AHR via JNK-dependent upregulation of bradykinin receptors.25) In addition, the present study revealed that DSP-induced bradykinin receptor-mediated airway contraction with an increased bradykinin receptor mRNA expression in the smooth muscle. This agrees well with our previous studies by immunohistochemistry using confocal microscopy seen that the upregulation of bradykinin receptors located in the airway smooth muscle.23,30)
Environmental cigarette smoke contains more than 5000 different substances, emission levels in mainstream smoke have been found for 542 of the components and a human inhalation risk value for 98 components.31) Nicotine is the major stimulant component inhaled from environmental cigarette smoke. It may exacerbate asthmatic AHR, airway inflammation, bacterial and viral infections. The DSP solution used in the present study contains 0.1 mg/mL nicotine. Previously, we showed that nicotine increased the expression of bradykinin B1 and B2 receptor-mediated airway contractions, but does not affect airway contractile responses to 5-HT, acetylcholine and endothelin receptor agonists.23) The present study has demonstrated that DSP could induce AHR in vivo, and upregulation of bradykinin B1 and B2 receptors was involved in DSP-induced AHR. It is possible that the effect of DSP may be at least partially mediated by nicotine.
The flexiVent™ system is the standard method for measuring lung functions of small animals such as mice1) and rats.32) In the present study, we have established a mouse in-vivo model of AHR by intranasal administration of smoke extracts DSP. After intranasal administration of different concentrations of DSP for 7 d, the mice exhibited AHR demonstrated by flexiVent™ system in vivo. We also employed an organ culture of mouse tracheal segments exposed to cigarette extract DSP with or without specific JNK SP600125 or the anti-inflammatory drug dexamethasone, and revealed that JNK-inflammatory signal pathways-mediated upregulation of bradykinin receptors was involved in cigarette smoke extract DSP-induced AHR in vitro. The in-vivo model with the flexiVent™ system is a standard method for examining small animal AHR, while the in-vitro organ culture model provides a simple and useful base for studying the molecular mechanisms involved in the pathogenesis of AHR. However, there are variations between the levels of receptor expression and the receptor activity in the present study. Protein expression is not only controlled by transcriptional mechanisms33) and thus mRNA expression poorly predicts the corresponding protein expression level. Furthermore, regulation of the receptor expression and functions involves more complex molecular mechanisms like the receptor-mediated contraction that involves phosphorylation of proteins34) and intracellular signaling cross-talks.35)
AHR is an important feature of asthma. Methacholine bronchial provocation by a 2-min tidal breathing method is highly sensitive; a PC20 value of less than 1 mg/mL/PD20 value of less than 25 µg is highly specific for AHR i.e., diagnostic, and a negative test result (PC20 > 16 mg/mL, PD20 > 400 µg) rules out current asthma.36) In murine models, flexiVent™ is often used to assess airway responsiveness to inhaled methacholine in various disease models including asthma, COPD, lung injury as well as airway inflammation.26) In the present study, DSP (0.75, 1.5 or 3 µL/mL) was administered intranasally daily for 7 d. At the end of this period, lung functions of the mice were measured with flexiVent™. The results showed that after intranasal treatment with different concentrations of DSP (0.75, 1.5 or 3 µL/mL) for 7 d, the mice exhibited significantly dose-dependent increases in the Rrs, as well as reduction of the Crs, demonstrating that DSP-induced AHR in mice in vivo.
Cigarette smoking is associated with accelerated decline of lung function, increased mortality, and worsening of symptoms in both asthma and COPD.37) It has been reported that in smokers the plasma concentration of nicotine is between 4–72 ng/mL while the average concentration is 33 ng/mL.38) The used concentration of DSP in the present study is supposed to be lower than plasma concentration of nicotine in smokers. The underlying molecular mechanisms behind how cigarette smoke induce AHR include increased expressions of endothelin receptors and 5-HT 2A receptors3,14) in airway smooth muscle cells, dysfunction of airway epithelium2) and nerve.1) The present study demonstrated that upregulation bradykinin receptors was involved in cigarette smoke extract DSP-induced AHR. This is supported by clinical studies showing that bradykinin receptor expressions are increased in the airway of patients with AHR39) and inhalation of bradykinin induces strong bronchial contraction in asthmatics.20) Our previous studies have suggested that cytokines upregulate bradykinin B1 and B2 receptors in airway smooth muscle cells by activating the JNK-mediated signalling pathway.25) Taken all together, this suggests that bradykinin receptor upregulation might play a key role in cigarette smoke associated AHR. In conclusion, the present study established an in-vivo murine model of AHR induced by cigarette smoke extract DSP and demonstrated that that DSP could induce AHR in vivo and in vitro. Furthermore, the upregulation of bradykinin receptors in airway is most likely one of the underlying molecular mechanisms that are responsible for cigarette smoke extract-induced AHR.
Acknowledgments
This study was supported by Grants from Shaanxi Province Department of Education (No. 17JS119 and No. 16JS097), Shaanxi Provincial Research Center for the Project of Prevention and Treatment of Respiratory Diseases (2017HXKF07), Xi’an Medical University (No. 2015RCYJ05).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Xu Y, Cardell LO. Long-term nicotine exposure dampens LPS-induced nerve-mediated airway hyperreactivity in murine airways. Am. J. Physiol. Lung Cell. Mol. Physiol., 313, L516–L523 (2017).
- 2) Lam DC, Kwok HH, Yu WC, Ko FW, Tam CY, Lau AC, Fong DY, Ip MS. CC16 levels correlate with cigarette smoke exposure in bronchial epithelial cells and with lung function decline in smokers. BMC Pulm. Med., 18, 47 (2018).
- 3) Xu CB, Lei Y, Chen Q, Pehrson C, Larsson L, Edvinsson L. Cigarette smoke extracts promote vascular smooth muscle cell proliferation and enhances contractile responses in the vasculature and airway. Basic Clin. Pharmacol. Toxicol., 107, 940–948 (2010).
- 4) Zhang Y, Cardell LO, Edvinsson L, Xu CB. MAPK/NF-kappaB-dependent upregulation of kinin receptors mediates airway hyperreactivity: a new perspective for the treatment. Pharmacol. Res., 71, 9–18 (2013).
- 5) Slats A, Taube C. Asthma an, d chronic obstructive pulmonary disease overlap: asthmatic chronic obstructive pulmonary disease or chronic obstructive asthma? Ther. Adv. Respir. Dis., 10, 57–71 (2016).
- 6) Tkacova R, Dai DLY, Vonk JM, Leung JM, Hiemstra PS, van den Berge M, Kunz L, Hollander Z, Tashkin D, Wise R, Connett J, Ng R, McManus B, Paul Man SF, Postma DS, Sin DD. Airway hyperresponsiveness in chronic obstructive pulmonary disease: a marker of asthma-chronic obstructive pulmonary disease overlap syndrome? J. Allergy Clin. Immunol., 138, 1571–1579.e10 (2016).
- 7) Hu G, Zhong N, Ran P. Air pollution and COPD in China. J. Thorac. Dis., 7, 59–66 (2015).
- 8) Drago G, Perrino C, Canepari S, Ruggieri S, L’Abbate L, Longo V, Colombo P, Frasca D, Balzan M, Cuttitta G, Scaccianoce G, Piva G, Bucchieri S, Melis M, Viegi G, Cibella F, Bilocca D, Borg C, Montefort S, Zammit C, Ferrante G, Grutta S, Melis MR, Minardi R, Ristagno R, Rizzo G. Relationship between domestic smoking and metals and rare earth elements concentration in indoor PM2.5. Environ. Res., 165, 71–80 (2018).
- 9) Xing YF, Xu YH, Shi MH, Lian YX. The impact of PM2.5 on the human respiratory system. J. Thorac. Dis., 8, E69–E74 (2016).
- 10) Loffredo CA, Tang Y, Momen M, Makambi K, Radwan GN, Aboul-Foutoh A. PM2.5 as a marker of exposure to tobacco smoke and other sources of particulate matter in Cairo, Egypt. Int. J. Tuberc. Lung Dis., 20, 417–422 (2016).
- 11) Aravamudan B, Thompson M, Sieck GC, Vassallo R, Pabelick CM, Prakash YS. Functional effects of cigarette smoke-induced changes in airway smooth muscle mitochondrial morphology. J. Cell. Physiol., 232, 1053–1068 (2017).
- 12) Garcia-Arcos I, Geraghty P, Baumlin N, Campos M, Dabo AJ, Jundi B, Cummins N, Eden E, Grosche A, Salathe M, Foronjy R. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax, 71, 1119–1129 (2016).
- 13) Shimoda T, Obase Y, Kishikawa R, Iwanaga T. Influence of cigarette smoking on airway inflammation and inhaled corticosteroid treatment in patients with asthma. Allergy Asthma Proc., 37, 50–58 (2016).
- 14) Cao L, Zhang Y, Cao YX, Edvinsson L, Xu CB. Secondhand smoke exposure causes bronchial hyperreactivity via transcriptionally upregulated endothelin and 5-hydroxytryptamine 2A receptors. PLOS ONE, 7, e44170 (2012).
- 15) Trifilieff A, Da Silva A, Gies JP. Kinins and respiratory tract diseases. Eur. Respir. J., 6, 576–587 (1993).
- 16) Ahluwalia A, Perretti M. B1 receptors as a new inflammatory target. Could this B the 1? Trends Pharmacol. Sci., 20, 100–104 (1999).
- 17) Barnes PJ. Bradykinin and asthma. Thorax, 47, 979–983 (1992).
- 18) Huang TJ, Haddad EB, Fox AJ, Salmon M, Jones C, Burgess G, Chung KF. Contribution of bradykinin B(1) and B(2) receptors in allergen-induced bronchial hyperresponsiveness. Am. J. Respir. Crit. Care Med., 160, 1717–1723 (1999).
- 19) Fuller RW, Dixon CM, Cuss FM, Barnes PJ. Bradykinin-induced bronchoconstriction in humans. Mode of action. Am. Rev. Respir. Dis., 135, 176–180 (1987).
- 20) Polosa R, Holgate ST. Comparative airway response to inhaled bradykinin, kallidin, and [des-Arg9]bradykinin in normal and asthmatic subjects. Am. Rev. Respir. Dis., 142, 1367–1371 (1990).
- 21) Ichinose M, Belvisi MG, Barnes PJ. Bradykinin-induced bronchoconstriction in guinea pig in vivo: role of neural mechanisms. J. Pharmacol. Exp. Ther., 253, 594–599 (1990).
- 22) Christiansen SC, Proud D, Sarnoff RB, Juergens U, Cochrane CG, Zuraw BL. Elevation of tissue kallikrein and kinin in the airways of asthmatic subjects after endobronchial allergen challenge. Am. Rev. Respir. Dis., 145, 900–905 (1992).
- 23) Xu Y, Zhang Y, Cardell LO. Nicotine enhances murine airway contractile responses to kinin receptor agonists via activation of JNK- and PDE4-related intracellular pathways. Respir. Res., 11, 13 (2010).
- 24) Xu CB, Zheng JP, Zhang W, Zhang Y, Edvinsson L. Lipid-soluble smoke particles upregulate vascular smooth muscle ETB receptors via activation of mitogen-activating protein kinases and NF-kappaB pathways. Toxicol. Sci., 106, 546–555 (2008).
- 25) Zhang Y, Adner M, Cardell LO. IL-1beta-induced transcriptional up-regulation of bradykinin B1 and B2 receptors in murine airways. Am. J. Respir. Cell Mol. Biol., 36, 697–705 (2007).
- 26) McGovern TK, Robichaud A, Fereydoonzad L, Schuessler TF, Martin JG. Evaluation of respiratory system mechanics in mice using the forced oscillation technique. J. Vis. Exp., 75, e50172 (2013).
- 27) Zhang Y, Adner M, Cardell LO. Up-regulation of bradykinin receptors in a murine in-vitro model of chronic airway inflammation. Eur. J. Pharmacol., 489, 117–126 (2004).
- 28) Keglowich LF, Borger P. The three a’s in asthma-airway smooth muscle, airway remodeling & angiogenesis. Open Respir. Med. J., 9, 70–80 (2015).
- 29) Zubakova R, Gille A, Faussner A, Hilgenfeldt U. Ca2+ signalling of kinins in cells expressing rat, mouse and human B1/B2-receptor. Int. Immunopharmacol., 8, 276–281 (2008).
- 30) Lei Y, Zhang Y, Cao Y, Edvinsson L, Xu CB. Up-regulation of bradykinin receptors in rat bronchi via I kappa B kinase-mediated inflammatory signaling pathway. Eur. J. Pharmacol., 634, 149–161 (2010).
- 31) Talhout R, Schulz T, Florek E, van Benthem J, Wester P, Opperhuizen A. Hazardous compounds in tobacco smoke. Int. J. Environ. Res. Public Health, 8, 613–628 (2011).
- 32) Kozma Rde L, Alves EM, Barbosa-de-Oliveira VA, Lopes FD, Guardia RC, Buzo HV, Faria CA, Yamashita C, Cavazzana M Junior, Frei F, Ribeiro-Paes MJ, Ribeiro-Paes JT. A new experimental model of cigarette smoke-induced emphysema in Wistar rats. J. Bras. Pneumol., 40, 46–54 (2014).
- 33) Foss EJ, Radulovic D, Shaffer SA, Goodlett DR, Kruglyak L, Bedalov A. Genetic variation shapes protein networks mainly through non-transcriptional mechanisms. PLoS Biol., 9, e1001144 (2011).
- 34) Luo G, Jamali R, Cao YX, Edvinsson L, Xu CB. Vascular endothelin ET(B) receptor-mediated contraction requires phosphorylation of ERK1/2 proteins. Eur. J. Pharmacol., 538, 124–131 (2006).
- 35) Bornfeldt KE, Krebs EG. Crosstalk between protein kinase A and growth factor receptor signaling pathways in arterial smooth muscle. Cell. Signal., 11, 465–477 (1999).
- 36) Nair P, Martin JG, Cockcroft DC, Dolovich M, Lemiere C, Boulet LP, O’Byrne PM. Airway hyperresponsiveness in asthma: measurement and clinical relevance. J. Allergy Clin. Immunol. Pract., 5, 649–659.e2 (2017).
- 37) Tamimi A, Serdarevic D, Hanania NA. The effects of cigarette smoke on airway inflammation in asthma and COPD: therapeutic implications. Respir. Med., 106, 319–328 (2012).
- 38) Russell MA, Jarvis M, Iyer R, Feyerabend C. Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. BMJ, 280, 972–976 (1980).
- 39) Christiansen SC, Eddleston J, Woessner KM, Chambers SS, Ye R, Pan ZK, Zuraw BL. Up-regulation of functional kinin B1 receptors in allergic airway inflammation. J. Immunol., 169, 2054–2060 (2002).