Abstract
Signal transducer and activator of transcription 3 (STAT3) is a latent transcription factor that contributes to tumor cell growth and survival and is often constitutively active in several types of cancers, which makes it an attractive target for cancer therapy. We identified 5,5′-(pentane-1,5′-diyl)bis(2-methyl-1,4-benzoquinone) (BPMB) as a new STAT3 inhibitor. BPMB inhibited the transcriptional activities of STAT3, despite its inability to reduce the phosphorylation and nuclear translocation of STAT3. BPMB selectively inhibited the proliferation of human breast cancer cell lines with constitutively activated STAT3. Furthermore, a gel retardation pattern was obtained by immunoblotting only when those STAT3-activated cell lines were treated with BPMB. The shifted bands could be immunoblotted with anti-STAT3 antibody but not with anti-STAT1/STAT5 antibody, and were stable under reducing conditions. The purified recombinant STAT3 protein treated with BPMB afforded a similar band shift pattern. Matrix-assisted laser desorption/ionization-mass spectrometry analysis of the component comprising the main shifted band suggested that the complex is a STAT3 homodimer crosslinked by BPMB through a Michael addition with Cys550 in the linker domain. Alanine replacement at this position resulted in reduction of the STAT3 dimer formation in the gel retardation assay. Thus, our results suggest that BPMB inhibits the proliferation of STAT3-activated cell lines, presumably through acylation of the linker domain and subsequent induction of the inactive STAT3 complexes.
INTRODUCTION
Signal transducers and activators of transcription (STATs) are cytoplasmic transcription factors that relay signals from growth factors and cytokines in the cytoplasm to the nucleus. The STAT family comprises seven members: STAT1–STAT4, STAT6, and two isoforms of STAT5, namely, STAT5a and STAT5b. Overexpression and/or constitutive activation of STAT3, in particular, have been detected in many types of hematopoietic and solid tumors, such as leukemia, and breast and prostate cancer.1–5) The STAT3 signaling pathway involves the activation of receptor tyrosine kinases, such as epidermal growth factor and platelet-derived growth factor receptors, and Janus kinases (JAKs). Following phosphorylation of the conserved Tyr705 residue in STAT3 (e.g., pYXXQ in the gp130 receptor for STAT3 binding),6) two STAT3 monomers dimerize through a reciprocal interaction between p-Tyr705 and the Src homology 2 (SH2) domains. This results in the translocation of activated STAT3 dimers into the nucleus, where they regulate gene expression by binding to specific DNA sequences.7–10) The mode of constitutive activation of STAT3 through posttranslational modifications (e.g., tyrosine and serine phosphorylation, lysine acetylation,11,12) and redox regulation13,14)) may vary in different types of cancers.
STAT3 plays a role in various biological functions, including cell proliferation, cell survival, apoptosis, and inflammation.15–17) The transcription of a number of genes involved in cell cycle progression, such as cyclin D1 and c-myc, as well as genes involved in angiogenesis (e.g., vascular endothelial growth factor) and anti-apoptosis (e.g., survivin, Bcl-2, and Bcl-xL) are activated by STAT3. It has been reported that STAT3 signaling restrains natural tumor immune surveillance and the inhibition of hematopoietic STAT3 in tumor-bearing hosts elicits multicomponent therapeutic antitumor immunity.18–20) It was also determined that in normal cells, blocking STAT3 is neither harmful nor toxic to the cells.7,21) The oncogenic functions of STAT3 and the therapeutic potential of its inhibition make the targeting of STAT3 an intriguing strategy in cancer treatment.
Several strategies have been examined to inhibit STAT3 activation, such as targeting the activating JAK2 kinase. Ruxolitinib22) and tofacitinib23) suppress the upper reaches of STAT signaling and induce apoptosis in tumor cells.24) However, JAK inhibitors target the ATP binding site, which is highly conserved across the known human protein kinases; thus, they are not STAT3-selective inhibitors. Therefore, there has been recent interest in STAT3-SH2 domain inhibitors and STAT3 DNA-binding inhibitors. Stattic and S3I-201 are STAT3-SH2 domain inhibitors designed to suppress the interaction between the SH2 domain and p-Tyr705.25,26) Galiellalactone and inS3-54 target the DNA binding domain of STAT3, thereby disrupting STAT3-DNA interactions.27,28) We previously identified STAT3 direct inhibitors, namely, 2-chloro-1,4-naphthalenedione analogue,29) 5,15-diphenylporphyrin,30) and STX-0119.31) However, only a limited number of STAT3-SH2 domain inhibitors and STAT3 DNA-binding inhibitors have reached pre-clinical and clinical trials. Several of these STAT3 inhibitors (e.g., stattic, galiellalactone, and S3I-201) were reported to be irreversible cysteine binders.32–34) Consequently, further development of irreversible STAT3 inhibitors and determination of the detailed mode of action are important for the identification of new STAT3 inhibitors.
In this study, we identified 5,5′-(pentane-1,5′-diyl)bis(2-methyl-1,4-benzoquinone) (BPMB) as a new STAT3 inhibitor. BPMB was previously synthesized by Hunig et al.35) as an intermediate of N,N′-dicyanoquinodiimine (DCNQI) copper salt analogues that exhibit metallic conductivity. However, to date, there has been no report of the biological activity of BPMB. We demonstrate here that BPMB inhibits the proliferation of STAT3-activated cells and abrogates the STAT3 function presumably by the induction of stable STAT3 complexes including a homodimer through a bifunctional intermolecular covalent reaction with Cys550 in the linker domain.
MATERIALS AND METHODS
ReagentsBPMB was purchased from Maybridge (U.K.). JAK inhibitor I was purchased from Sigma-Aldrich (U.S.A.). Anti-STAT3, anti-STAT1, anti-STAT5, anti-phospho STAT3 (Tyr705), and anti-phospho STAT3 (Ser727) antibodies were obtained from Cell Signaling Technology (U.S.A.). Anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was purchased from Abcam (U.K.). Horseradish peroxidase (HRP)-conjugated anti-rabbit immunoglobulin G (IgG), anti-mouse IgG, and anti-rabbit IgG were purchased from GE Healthcare Biosciences (U.S.A.). Streptavidin-HRP was obtained from Amersham Biosciences (U.K.). Onscostatin M (OSM) and interferon-γ (IFN-γ) was obtained from Wako (Japan) and Sigma-Aldrich, respectively. Escherichia coli, strain BL-21 Star (DE3) pLysS, was purchased from Life Technologies Corporation (U.S.A.). pBluescript II KS (+) was bought from Stratagene (U.S.A.) and pET-28a (+) was obtained from Novogene (China). PhosphoSafe Extraction Reagent was purchased from Merck (U.S.A.), and Protein G Plus /Protein A Agarose Suspension was obtained from EMD Millipore (U.S.A.). The other reagents were purchased from Nacalai Tesque (Japan).
Cell Lines and CultureThe STAT3 and STAT1 reporter HeLa stable cell lines for the luciferase reporter gene assay were purchased from Signosis (U.S.A.) (product No. SL-0003 and SL-0004, respectively). Cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) in addition to 10% (v/v) fetal bovine serum (FBS), 20 U/mL penicillin, 20 µg/mL streptomycin, and 100 µg/mL hygromycin B at 37°C in a humidified atmosphere with 5% CO2. HeLa cells (cervical carcinoma) were obtained from RIKEN BRC (Japan). The cells were maintained in DMEM supplemented with 10% (v/v) FBS, 20 U/mL penicillin, and 20 µg/mL streptomycin at 37°C in a humidified atmosphere with 5% CO2. MDA-MB-231, MDA-MB-453, MDA-MB-468, and MCF7 cells (breast cancer) were purchased from the American Type Culture Collection (U.S.A.). MDA-MB-231, MDA-MB-453, and MDA-MB-468 cells were grown in DMEM/Ham’s F-12 with 10% (v/v) FBS, 20 U/mL penicillin, 20 µg/mL streptomycin, and 100 µg/mL hygromycin B at 37°C in a humidified atmosphere with 5% CO2. MCF7 cells were maintained in DMEM supplemented with 5% (v/v) FBS, 40 U/mL penicillin, and 40 µg/mL streptomycin at 37°C in a humidified atmosphere with 5% CO2.
Plasmid Construction and Protein ExpressionFull-length human STAT3 cDNA was obtained from a human cDNA library using PCR and subcloned into pBluescript II KS (+). The nucleotides coding for human STAT3 were separately cloned into the BamHI/Sall sites of modified pET-28a (+) and 6 × His-tag and Avi-tag were introduced at the N-terminus of the protein. E. coli BirA-BL-21 competent cells transfected with the constructed pET-28a (+) plasmid were grown in LB medium containing 10 µg/mL of chloramphenicol and kanamycin at 37°C to an OD600 of 0.4. The cells were induced with 1 mM of isopropyl-β-D-thiogalactopyranoside and 40 µM biotin for 3 h at 25°C. Cells were centrifuged at 5000 rpm for 10 min at 4°C after cooling for 10 min. The bacterial pellets were suspended in lysis buffer (0.1% (w/v) sucrose, 40 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES)–KOH (pH 7.9), 500 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.5% (v/v) NP-40, 1 mM dithiothreitol (DTT), and 1 Tab/50 mL complete protease inhibitor cocktail tablets). The cells were lysed with 10 cycles of sonication, each consisting of a constant pulse for a 1 min on ice. Cell lysates were centrifuged at 15000 rpm for 15 min at 4°C. The supernatants were loaded onto a Ni Sepharose High Performance column for 90 min at 4°C. Proteins that had non-specifically bound to the column were washed out with wash buffer (1% (w/v) sucrose, 40 mM Hepes–KOH (pH 7.9), 300 mM NaCl, 1 mM EDTA, 1 mM DTT, 0.3% (v/v) Brij-35, and 1 Tab/50 mL protease inhibitor cocktail tablets). Recombinant proteins were eluted with elution buffer (1% (w/v) sucrose, 80 mM Hepes–KOH (pH 7.9), 600 mM NaCl, 2 mM EDTA, 0.02% (v/v) NP-40, 10% (v/v) glycerol, and 0.2 mM phenylmethylsulfonyl fluoride (PMSF)). Imidazole in the elution fraction was removed with dialysis buffer (10 mM Hepes–KOH (pH 7.4), 50 mM NaCl, 10 mM β-mercaptoethanol, 0.1 mM ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA), 0.02% (w/v) NP-40, 0.2 mM PMSF, and 10% (v/v) glycerol) at 4°C overnight. The purified proteins were snap-frozen in liquid nitrogen and stored at −80°C before use. Point mutations were inserted into rhSTAT3 by using inverse PCR technology. Cys550 to alanine was mutated. Inverse PCR was performed by using pBluescript II KS (+) as a template via KOD-Plus-ver. 2 (Toyobo, Japan). The oligonucleotide primer sets were: Cys550 (forward: 5′-GCT AAA TTT GCC AAA GAA AAC ATG GCT GGC-3′, reverse: 5′-CCA TGT GAT CTG ACA CCC TGA ATA ATT CA CAC C-3′). Inverse PCR (1 × PCR buffer, 15 pmol 5′ primer and 3′ primer, 0.2 mM dNTPs, 50 ng STAT3 cDNA, 1U KOD Plus, and 1.5 mM MgSO4) was performed for 2 min at 94°C, (10 s at 98°C and 6.5 min at 68°C) for 7 cycles. The amplified fragment was ligated by using Ligation High ver. 2 (Toyobo), and the resultant plasmid was cloned into the HindIII/XhoI sites modified pET-28a (+).
Luciferase Reporter Gene AssayFor the STAT3/STAT1-dependent reporter assay, the cells (2 × 104 cells/well) were seeded into white 96-well plates (Corning, U.S.A.) and incubated overnight at 37°C in 5% CO2. The cells were treated with vehicle or test compounds for 1 h. The cells were then stimulated with 10 ng/mL OSM (STAT3) or 6 ng/mL IFN-γ (STAT1) for 4 h. The medium was aspirated off and Steady-Glo (Promega) was added to the cells. The plate was then placed on a shaker 6 for 10 min. Luminescence was detected using an ARVO Light plate reader (PerkinElmer, Inc., U.S.A.). The relative signal intensity was calculated in each well as the ratio for the mean signal of the vehicle.
Western Blot AnalysisThe samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and stained with Imperial Protein Stain (Thermo Fisher Scientific, U.S.A.). For Western blotting, the proteins were transferred onto a polyvinylidene fluoride membrane. The membranes were incubated with blocking buffer (5% (w/v) skim milk in Tris-buffered saline (pH 7.4) and 0.1% (v/v) Tween 20) overnight at 4°C. They were then incubated with primary antibody diluted in blocking buffer overnight at 4°C. Thereafter, they were incubated with HRP-conjugated secondary antibodies diluted in blocking buffer for 1 h at room temperature. Immunoblots were developed by using a chemiluminescent substrate and detected using a LAS-3000 imager (GE Healthcare).
Nuclear Translocation AssayThe Cellomics STAT3 Activation Kit (Thermo Scientific) was used for the nuclear translocation assay. HeLa cells (1 × 104 cells/well) were incubated in a 96-well microplate for 24 h. The cells were pretreated with vehicle or test compounds for 1 h, and 30 ng/mL OSM was applied and the mixture was incubated for 10 min. The subsequent procedures were performed according to the manufacturer’s instructions. In brief, fixation solution was applied and incubation for 10 min. The cells were incubated with permeabilization buffer for 15 min and were incubated with the primary antibody for 1 h, followed by secondary antibody for 1 h and then incubated with 10% Vectashield (H-1000). The cells were scanned with an ArrayScan VTI reader (Thermo Scientific), and analyzed with the Cytoplasm to Nucleus Translocation Application. The mean fluorescent values in the nuclei were calculated. The signals for vehicle treatment and no treatment (without oncostatin M) were represented as 100 and 0%, respectively, and the relative signal intensity was also calculated.
Gel Retardation AssayWhole-cell lysates prepared from OSM-stimulated cells or the purified rhSTAT3 protein were incubated with vehicle or BPMB for 1 h at 4°C. The samples were subjected to SDS-PAGE and subsequent blotting with streptavidin–HRP conjugate.
ImmunoprecipitationCells were treated vehicle or BPMB for 1 h and lysed in PhosphoSafe extraction reagent. The lysates were precleared with a mixture of Protein G Plus/Protein A agarose suspension for 20 min at 4°C. Bead pellets were discarded by centrifugation at 15000 rpm for 5 min and the supernatant was retained for immunoprecipitation. The lysates were immunoprecipitated with anti-STAT3 antibody for 4 h at 4°C on a rotator. The immunocomplexes were captured by the addition of a Protein G Plus/Protein A Agarose Suspension slurry overnight at 4°C. Samples were rinsed three times with lysis buffer and equal amounts of protein were subjected to SDS-PAGE and subsequent Western blotting.
Mass Spectrometry Analysis of the Adduct of BPMB with rhSTAT3Samples were subjected to SDS-PAGE and stained with Imperial Protein Stain. The component of the main shifted band was decolored with reagents containing 50 mM ammonium bicarbonate and 50% (v/v) acetonitrile and then reduced in the gel with 100 mM DTT in 80 mM ammonium bicarbonate for 30 min at 56°C. The gel pieces were incubated with 100 mM iodoacetamide in 80 mM ammonium bicarbonate for 45 min with shaking at room temperature and then digested by 0.01 µg/µL trypsin for 16 h at 37°C. Digestion was stopped by the addition of 0.1% (v/v) trifluoroacetic acid in 50% (v/v) acetonitrile. Tryptic peptides were extracted and 1 µL was mixed with an equal volume of 3 g/L 5-dihydroxybenzoic acid and 0.1% (v/v) trifluoroacetic acid in 30% (v/v) acetonitrile as a matrix, loaded onto the target plate, and air-dried. The samples were analyzed by ultrafleXtreme (Bruker Daltonics, U.S.A.) in the positive mode. The data were processed by Mascot Distiller searching the NCBI RefSeq human database.
WST-8 AssayCells were plated in a 96-well tissue culture plate and incubated for 24 h. Cells were treated with vehicle or BPMB (0.1–3 µM) for 72 h. After incubation, WST-8 solution (Dojindo Molecular Technologies, U.S.A.) was added to each well and incubated at 37°C for 4 h, and the absorbance was read at 450 nm.
Statistical AnalysisAll experiments were performed in triplicate. In the nuclear translocation assay, the statistical significance was calculated by Student’s t-test with * p < 0.05 and ** p < 0.01 versus vehicle treatment. In the luciferase reporter gene assay, statistical significance was calculated by Student’s t-test with * p < 0.05 and ** p < 0.01 versus luc-STAT3. In the WST-8 assay, statistical significance was calculated by one-way ANOVA followed by Tukey’s multiple comparisons test.
RESULTS
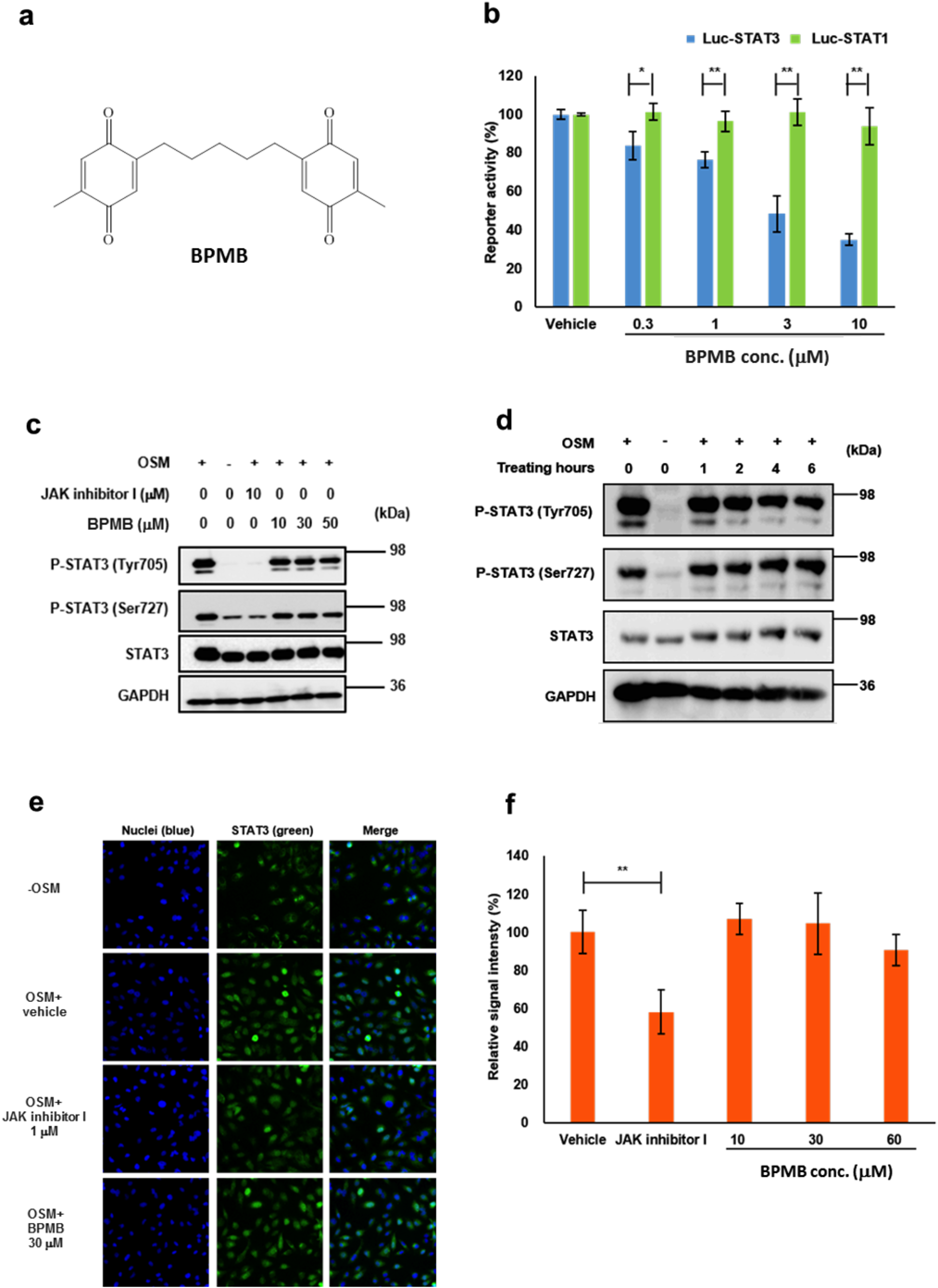
BPMB Inhibited Transcriptional Activation of STAT3 without Affecting Its Phosphorylation Status and Nuclear TranslocationIn our screening campaign for STAT3 inhibitors, we identified BPMB as a hit compound by its ability to inhibit STAT3-transcriptional activity (Fig. 1a). As shown in Fig. 1b, BPMB inhibited STAT3-dependent transcriptional activity (half maximal inhibitory concentration (IC50) = 3.57 µM) but not STAT1-dependent transcriptional activity (IC50 > 10 µM). To verify that BPMB inhibited STAT3 activation, we examined its effect on the phosphorylation of STAT3 in OSM-stimulated HeLa cells (Figs. 1c, d). Phosphorylation of STAT3 was observed in HeLa cells stimulated with OSM and treatment with increasing concentrations of BPMB rarely affected the phosphorylation status of either Tyr705 or Ser727.
We investigated the effect of BPMB on STAT3 nuclear translocation by immunostaining. As shown in Figs. 1e and f, nuclear translocation of STAT3 was observed within 10 min in HeLa cells stimulated with OSM. The addition of BPMB up to 60 µM resulted in no significant reduction in nuclear STAT3. These results suggest that BPMB may directly inhibit STAT3 without affecting the upstream regulators or nuclear import factors.
Inhibition of Proliferation and Induction of STAT3 Complexes by BPMB in STAT3-Activated Cancer CellsWe examined whether BPMB could inhibit proliferation in human breast cancer cells with constitutively activated STAT3. Constitutive phosphorylation of STAT3 was observed in MDA-MB-231 and MDA-MB-468 cells, but not in MDA-MB-453 or MCF7 cells (Fig. 2b). The cells were treated with 0.1–3 µM BPMB for 72 h and assessed using the WST-8 cell proliferation assay. As shown in Fig. 2a and Supplementary Table 1, BPMB significantly suppressed the proliferation of MDA-MB-231 and MDA-MB-468 cells in a dose-dependent manner, but did not affect MDA-MB-453 or MCF7 cells. Those results demonstrate that BPMB selectively inhibits the proliferation of cell lines with constitutively activated STAT3. Interestingly, we observed unusual retarded bands with masses corresponding to around 140–250 kDa in immunoprecipitates from cell lysates of MDA-MB-231 and MDA-MB-468 cells treated with BPMB (Fig. 2b). However, retarded bands were not detected in MDA-MB-453 or MCF7 cells. The components of these high-molecular weight bands were not separable on SDS-PAGE and were recognized by the anti-STAT3 antibody. They are, therefore, SDS-resistant protein complexes harboring STAT3.
Characterization of the STAT3-Containing Complexes Induced by BPMBThe formation of STAT3/1 and STAT3/5 heterodimers has been reported to be induced by several cytokines.36) To examine whether the complexes induced by BPMB contain STAT1 and/or STAT5, we analyzed the component immunoprecipitated with an anti-STAT3 antibody from HeLa cell lysates. The immunoprecipitates from BPMB-treated cell lysates were immunoblotted with anti-STAT3, anti-STAT1, or anti-STAT5 antibodies. A similar gel retardation pattern was only detectable by the anti-STAT3 antibody in HeLa cells stimulated with OSM (Fig. 3a), suggesting that BPMB preferentially interacted with Tyr-phosphorylated STAT3 in cells and the large complexes could be STAT3 homooligomers or complexes with other STAT3 binding partners.
A previous study reported that H2O2 generates STAT3 oligomers.13) We attempted to understand the differences between STAT3 complexes formed by exposure to H2O2 or BPMB. To this end, the STAT3 complexes induced by H2O2 or BPMB were pre-treated with a reducing agent in lysates from OSM-stimulated cells before PAGE-separation and immunoblotting. The gel retardation pattern by exposure to H2O2 and BPMB was also detected in in vitro experiments. Although the band corresponding to the STAT3 oligomers formed by H2O2 collapsed, the bands corresponding to STAT3 complexes formed by BPMB were not labile in the presence of 100 mM DTT (Fig. 3b). Additionally, the mobilities of the STAT3 complexes within the gel differed between those formed by H2O2 and those by BPMB. This observation indicated that BPMB induced covalently-linked STAT3 complexes that were different from H2O2-induced disulfide-linked STAT3 oligomers.
Modification of Cys550 within the Linker Domain of STAT3 by BPMBBPMB contains two quinoid moieties in the structure (Fig. 1a). Based on the chemical reactivity of the quinoid moieties, we predicted that BPMB could modify STAT3 proteins through alkylation and thereby inhibit the transcriptional activity of STAT3. To gain more insight into the biochemical interaction, we examined the effect of BPMB on purified rhSTAT3 in vitro. Biotinylated rhSTAT3 purified by His-tag dependent Ni-affinity chromatography was used for further biochemical analysis. Incubation of BPMB with rhSTAT3 for 1 h on ice resulted in the generation of the shifted bands migrating in the range 140–250 kDa, as detected by an avidin-HRP conjugate for biotin-based detection (Fig. 4a). We performed matrix-assisted laser desorption/ionization-mass spectrometry (MALDI-MS) analysis of the proteins in the shifted band that BPMB presumably had alkylated the cysteine residue(s) on STAT3. The gel piece containing the main band was dissolved in ammonium bicarbonate and reduced with 100 mM DTT, and then digested with trypsin, and the resulting peptide mixture was subjected to MALDI-MS analysis. Here, we defined peptide X as the peptide fragment modified by BPMB. A mass signal at m/z 6057.85 was detected only in STAT3 peptides treated with BPMB and could not be assigned to any peptide within the native STAT3 sequence, making this peptide, [peptide X-BPMB-peptide X-H+], a putative candidate for BPMB modification (Fig. 4b). There are 199 theoretical STAT3 tryptic peptide fragments in total. We calculated the theoretical weight of the two STAT3 tryptic peptide fragments to which BPMB was bifunctionally bound (Supplementary Fig. 1, Supplementary Table 2). The peptide corresponding to amino acids 532–557 of STAT3, with a theoretical weight of 6057.93, was consistent with the observed weight of 6057.85 found by MALDI-MS, thereby indicating that [peptide X] was the 532–557 peptide [LLGPGVNYSGCQITWAKFCKENMAGK] and contained cysteine residues at positions 542 and 550. Cys542 was subjected to alkylation by iodoacetamide (Supplementary Table 3). The results indicated that the shifted main band corresponded to STAT3 homodimer crosslinked by BPMB through a bifunctional covalent reaction with Cys550 in the linker domain of STAT3 (Fig. 4c). Our MALDI-MS analysis suggested that Cys550 could be an important primary target cysteine on STAT3. To better understand the role of Cys550, we assessed the effect of BPMB on a recombinant STAT3 protein with an alanine replacing Cys550. This mutant recombinant STAT3 was prepared and evaluated using the gel retardation assay. In the presence of 1 µM BPMB, the intensities of the retarded bands were decreased for the C550A mutant (Fig. 4d). These results demonstrated that Cys550 is an important residue for the formation of STAT3 homodimer crosslinked by BPMB in vitro.
DISCUSSION
Drugs that covalently modify their targets are rarely considered when attempting target-directed drug discovery, largely due to safety concerns and off-target reactivity problems. However, irreversible inhibitors such as ibrutinib,37) neratinib,38) and osimertinib39) have recently been widely used and studied because of their numerous advantages, including increased biochemical efficacy, longer duration of action, and the potential to inhibit processes that are often drug resistant. Several thiol modifiers have been reported to inhibit STAT3 functions, and one of these, namely, stattic, alkylates Cys251, Cys259, Cys367, and Cys426 in unphosphorylated STAT3.32) Moreover, galiellalactone binds to Cys367, Cys468, and Cys542 in unphosphorylated STAT333) and C48 (NSC-368262) modified Cys468.40) The C-28 methyl ester of the oleane triterpenoid 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO-Me) inhibits STAT3 dimerization by binding to Cys259.41) S3I-201 modifies five cysteines (Cys108, Cys259, Cys367, Cys542, and Cys687) in the full-length STAT3 protein.34) Furthermore, curcumin interacts directly with Cys259.42)
In this report, we identified BPMB as a novel STAT3 inhibitor due to its ability to inhibit STAT3 dependent transcriptional activity in the luciferase reporter assay (Fig. 1b). BPMB contains two quinoid moieties for acting as Michael acceptors (Fig. 1a). MALDI-MS analysis suggested that BPMB undergoes a Michael reaction with Cys550, generating a crosslinked STAT3 homodimer (Fig. 4c). Indeed, the Cys550 mutant was insusceptible to BPMB modification as assessed by the gel retardation assay (Fig. 4d). The Cys550 residue is located within STAT3 linker domain and has been reported to be involved in several functions such as STAT3 DNA binding, transcriptional activity, nuclear export, and protein–protein interactions.43) Taken together, Cys550 is likely a key residue for the intermolecular crosslinking of STAT3 followed by the inhibition of STAT3 function by BPMB. The reason why BPMB selectively alkylates Cys550 remains to be elucidated. The proximity effect in a reaction environment would possibly assist the Michael addition. To date, there have been no reports of Cys550-interacting STAT3 inhibitors. Thus, our findings suggest that Cys550 could be one of the important drug targets for the future development of new irreversible STAT3 inhibitors. However, we could still not exclude the possibility of the modification of other cysteines of STAT3 by BPMB.
Sobotta et al. reported that a H2O2-inducing redox relay generates disulfide-linked STAT3 dimers and tetramers with attenuated transcriptional activity.13) The STAT3-containing large complexes induced by BPMB were SDS-resistant and stable under reducing conditions, unlike the labile complexes induced by H2O2 (Fig. 3b). These results support the BPMB-mediated covalent linkage of STAT3 and exclude the possibility of the disulfide linkages in STAT3 complexes. As reported previously, H2O2 induces the formation of STAT3 oligomers in the absence of STAT3-activating cytokines.13) On the other hand, the BPMB-induced STAT3 complexes could be detected only by using cells with high levels of P-STAT3 (Figs. 2b, 3a). As shown in Fig. 3a, there was negligible contamination with STAT3/STAT1 or STAT3/STAT5 hetero-complex; thus, the large complexes are likely STAT3 dimers and tetramers covalently crosslinked with BPMB. Alternatively, they might be hetero-complexes of phosphorylated STAT3 with other unknown binding partners. We demonstrated that BPMB attenuated STAT3-dependent transcription in the luciferase reporter assay in HeLa cells stimulated with OSM, despite no effect to phosphorylation status or nuclear translocation (Fig. 1). The STAT3-containing complexes induced by BPMB were detected only in cell lines with high levels of Tyr-phosphorylated STAT3 (Fig. 2b). In addition, BPMB selectively inhibited the proliferation of cancer cell lines with high levels of Tyr-phosphorylated STAT3 (Fig. 2a). These results support the hypothesis that the STAT3-containing complexes were no longer functional and were unable to activate the transcription of target genes. Our results to date have partially revealed the mechanism by which BPMB inhibits STAT3. The mechanisms underlying BPMB-mediated STAT3 inhibition are possibly more complex than those described in this study, thus further investigation will be necessary to elucidate the precise mechanisms, especially in cellular systems.
In conclusion, we identified a benzoquinone derivative BPMB as a direct inhibitor of STAT3 and demonstrated its unique mode of action to form stable STAT3 complexes in cells. The Cys550 residue within the linker domain is likely critical for the inhibition of STAT3 function. Thus, BPMB might be a useful tool for designing the next generation of STAT3 inhibitors.
Acknowledgments
This work was supported by a JSPS Grant-in-Aid for Scientific Research (26430166).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Bowman T, Garcia R, Turkson J, Jove R. STAT in oncogenesis. Oncogene, 19, 2474–2488 (2000).
- 2) Turkson J, Jove R. STAT protein: novel molecular targets for cancer drug discovery. Oncogene, 19, 6613–6626 (2000).
- 3) Benekli M, Baer MR, Baumann H, Wetzler M. Signal transducer and activator of transcription proteins in leukemias. Blood, 101, 2940–2954 (2003).
- 4) Yu H, Jove R. The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer, 4, 97–105 (2004).
- 5) Turkson J. STAT proteins as novel targets for cancer drug discovery. Expert Opin. Ther. Targets, 8, 409–422 (2004).
- 6) Lamb P, Tapley P, Rosen J. Biochemical approaches to discovering modulators of the JAK-STAT pathway. Drug Discov. Today, 3, 122–130 (1998).
- 7) Buettner R, Mora LB, Jove R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res., 8, 945–954 (2002).
- 8) Desrivierès S, Kunz C, Barash I, Vafaizadeh V, Borghouts C, Groner B. The biological functions of the versatile transcription factors STAT3 and STAT5 and new strategies for their targeted inhibition. J. Mammary Gland Biol. Neoplasia, 11, 75–87 (2006).
- 9) Klampfer L. Signal transducers and activators of transcription (STATs): novel targets of chemopreventive and chemotherapeutic drugs. Curr. Cancer Drug Targets, 6, 107–121 (2006).
- 10) Haura EB, Turkson J, Jove R. Mechanisms of disease: Insights into the emerging role of signal transducers and activators of transcription in cancer. Nat. Clin. Pract. Oncol., 2, 315–324 (2005).
- 11) O’Shea JJ, Kanno Y, Chen X, Levy D. Cell signaling. Stat acetylation–a key facet of cytokine signaling? Science, 307, 217–218 (2005).
- 12) Yuan ZL, Guan YJ, Chatterjee D, Chin YE. STAT3 dimerization regulated by reversible acetylation of a single lysine residue. Science, 307, 269–273 (2005).
- 13) Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T, Scharf AN, Dick TP. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol., 11, 64–70 (2015).
- 14) Li L, Shaw PEA. STAT3 dimer formed by inter-chain disulphide bridging during oxidative stress. Biochem. Biophys. Res. Commun., 322, 1005–1011 (2004).
- 15) Peyser ND, Grandis JR. Critical analysis of the potential for targeting STAT3 in human malignancy. Onco Targets Ther., 6, 999–1010 (2013).
- 16) Sellier H, Re’billard A, Guette C, Barre´ B, Coqueret O. How should we define STAT3 as an oncogene and as a potential target for therapy? JAK-STAT, 2, e24716 (2013).
- 17) Quintás-Cardama A, Verstovsek S. Molecular pathways: JAK/STAT pathway: mutations, inhibitors, and resistance. Clin. Cancer Res., 19, 1933–1940 (2013).
- 18) Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mulé J, Kerr WG, Jove R, Pardoll D, Yu H. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med., 11, 1314–1321 (2005).
- 19) Buettner R, Huang M, Gritsko T, Karras J, Enkemann S, Mesa T, Nam S, Yu H, Jove R. Activated signal transducers and activators of transcription 3 signaling induces CD46 expression and protects human cancer cells from complement-dependent cytotoxicity. Mol. Cancer Res., 5, 823–832 (2007).
- 20) Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol., 7, 41–51 (2007).
- 21) Inghirami G, Chiarle R, Simmons WJ, Piva R, Schlessinger K, Levy DE. New and old functions of STAT3: a pivotal target for individualized treatment of cancer. Cell Cycle, 4, 1131–1133 (2005).
- 22) Mesa RA. Ruxolitiib, a selective JAK1 and JAK2 inhibitor for the treatment of myeloproliferative neoplasm and psoriasis. IDrugs, 13, 394–403 (2010).
- 23) Flanagan ME, Blumenkopf TA, Brissette WH, Brown MF, Casavant JM, Shang-Poa C, Doty JL, Elliott EA, Fisher MB, Hines M, Kent C, Kudlacz EM, Lillie BM, Magnuson KS, McCurdy SP, Munchhof MJ, Perry BD, Sawyer PS, Strelevitz TJ, Subramanyam C, Sun J, Whipple DA, Changelian PS. Discovery of CP-690, 550: a potent and selective Janus kinase (JAK) inhibitor for the treatment of autoimmune diseases and organ transplant rejection. J. Med. Chem., 53, 8468–8484 (2010).
- 24) Pedranzini L, Dechow T, Berishaj M, Comenzo R, Zhou P, Azare J, Bornmann W, Bromberg J. Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res., 66, 9714–9721 (2006).
- 25) Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. U.S.A., 104, 7391–7396 (2007).
- 26) Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol., 13, 1235–1242 (2006).
- 27) Huang W, Dong Z, Wang F, Peng H, Liu JY, Zhang JT. A small molecule compound targeting STAT3 DNA-binding domain inhibits cancer cell proliferation, migration, and invasion. ACS Chem. Biol., 9, 1188–1196 (2014).
- 28) Hellsten R, Johansson M, Dahlman A, Dizeyi N, Sterner O, Bjartell A. Galiellalactone is a novel therapeutic candidate against hormone-refractory prostate cancer expressing activated Stat3. Prostate, 68, 269–280 (2008).
- 29) Takakuma K, Ogo N, Uehara Y, Takahashi S, Miyoshi N, Asai A. Novel multiplexed assay for identifying SH2 domain antagonists of STAT family proteins. PLOS ONE, 8, e71646 (2013).
- 30) Uehara Y, Mochizuki M, Matsuno K, Haino T, Asai A. Novel high-throughput screening system for identifying STAT3-SH2 antagonists. Biochem. Biophys. Res. Commun., 380, 627–631 (2009).
- 31) Matsuno K, Masuda Y, Uehara Y, Sato H, Muroya A, Takahashi O, Yokotagawa T, Furuya T, Okawara T, Otsuka M, Ogo N, Ashizawa T, Oshita C, Tai S, Ishii H, Akiyama Y, Asai A. Identification of a new series of STAT3 inhibitors by virtual screening. ACS Med. Chem. Lett, 1, 371–375 (2010).
- 32) Heidelberger S, Zinzalla G, Antonow D, Essex S, Basu BP, Palmer J, Husby J, Jackson PJ, Rahman KM, Wilderspin AF, Zloh M, Thurston DE. Investigation of the protein alkylation sites of the STAT3:STAT3 inhibitor Stattic by mass spectrometry. Bioorg. Med. Chem. Lett., 23, 4719–4722 (2013).
- 33) Don-Doncow N, Escobar Z, Johansson M, Kjellstrom S, Garcia V, Munoz E, Sterner O, Bjartell A, Hellsten R. Galiellalactone is a direct inhibitor of the transcription factor STAT3 in prostate cancer cells. J. Biol. Chem., 289, 15969–15978 (2014).
- 34) Ball DP, Lewis AM, Williams D, Resetca D, Wilson DJ, Gunning PT. Signal transducer and activator of transcription 3 (STAT3) inhibitor, S3I-201, acts as a potent and non-selective alkylating agent. Oncotarget, 7, 20669–20679 (2016).
- 35) Siegfried H, Klaus S, Martina K, Uwe L, Harald R, Svante S, Jost-Ulrich von S, Hans-Christoph W. Tethered 1,4-benzoquinones and their DCNQI derivatives: syntheses, electronic interactions, redox properties, charge–transfer complexes, and copper salts. Eur. J. Org. Chem., 9, 1977–1988 (1998).
- 36) Delgoffe GM, Vignali DA. STAT heterodimers in immunity: a mixed message or a unique signal? JAK-STAT, 2, e23060 (2013).
- 37) Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, Hessler JD, Liu TM, Chang BY, Larkin KM, Stefanovski MR, Chappell DL, Frissora FW, Smith LL, Smucker KA, Flynn JM, Jones JA, Andritsos LA, Maddocks K, Lehman AM, Furman R, Sharman J, Mishra A, Caligiuri MA, Satoskar AR, Buggy JJ, Muthusamy N, Johnson AJ, Byrd JC. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood, 122, 2539–2549 (2013).
- 38) Rabindran SK, Discafani CM, Rosfjord EC, Baxter M, Floyd MB, Golas J, Hallett WA, Johnson BD, Nilakantan R, Overbeek E, Reich MF, Shen R, Shi X, Tsou HR, Wang YF, Wissner A. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res., 64, 3958–3965 (2004).
- 39) Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov., 4, 1046–1061 (2014).
- 40) Buettner R, Corzano R, Rashid R, Lin J, Senthil M, Hedvat M, Schroeder A, Mao A, Herrmann A, Yim J, Li H, Yuan YC, Yakushijin K, Yakushijin F, Vaidehi N, Moore R, Gugiu G, Lee TD, Yip R, Chen Y, Jove R, Horne D, Williams JC. Alkylation of cysteine 468 in Stat3 defines a novel site for therapeutic development. ACS Chem. Biol., 6, 432–443 (2011).
- 41) Ahmad R, Raina D, Meyer C, Kufe D. Triterpenoid CDDO-methyl ester inhibits the Janus-activated kinase-1 (JAK1) signal transducer and activator of transcription-3 (STAT3) pathway by direct inhibition of JAK1 and STAT3. Cancer Res., 68, 2920–2926 (2008).
- 42) Hahn YI, Kim SJ, Choi BY, Cho KC, Bandu R, Kim KP, Kim DH, Kim W, Park JS, Han BW, Lee J, Na HK, Cha YN, Surh YJ. Curcumin interacts directly with the Cysteine 259 residue of STAT3 and induces apoptosis in H-Ras transformed human mammary epithelial cells. Sci. Rep., 8, 6409 (2018).
- 43) Lim CP, Cao X. Structure, function, and regulation of STAT proteins. Mol. Biosyst., 2, 536–550 (2006).