Abstract
We previously demonstrated that Bacopa monnier (L.) WETTST. extract (BME) ameliorated cognitive dysfunction in animal models of dementia by enhancing synaptic plasticity-related signaling in the hippocampus and protecting cholinergic neurons in the medial septum. To further clarify the pharmacological features and availability of BME as a novel anti-dementia agent, we investigated whether BME affects neuronal repair using a mouse model of trimethyltin (TMT)-induced neuronal loss/self-repair in the hippocampus. Mice pretreated with TMT (2.8 mg/kg, intraperitoneally (i.p.)) on day 0 were given BME (50 mg/kg, per os (p.o.)) once daily for 15–30 d. Cognitive performance of the animals was elucidated twice by the object location test and modified Y maze test on days 17–20 (Phase I) and days 32–35 (Phase II) or by the passive avoidance test on Phase II. TMT impaired hippocampus-dependent spatial working memory and amygdala-dependent fear-motivated memory. The administration of BME significantly prevented TMT-induced cognitive deficits. The protective effects of BME on the spatial memory deficits were confirmed by Nissl staining of hippocampal tissues and propidium iodide staining of organotypic hippocampal slice cultures. Immunohistochemical studies conducted on days 17 and 32 revealed that thirty days of treatment with BME increased the number of 5-bromo-2′-deoxyuridine (BrdU)-immunopositive cells in the dentate gyrus region of TMT-treated mice, whereas fifteen days of treatment with BME had no effect. These results suggest that BME ameliorates TMT-induced cognition dysfunction mainly via protecting the hippocampal neurons from TMT-induced hippocampal lesions and partly via promoting neuroregeneration in the dentate gyrus regions.
INTRODUCTION
Bacopa monnieri (L.) WETTST. (BM) is a plant belonging to the family Scrophulariaceae and is well known as a medicinal plant traditionally used in India and other Southeast Asian countries to enhance memory and improve brain function. Previous studies using various animal models of cognitive deficits demonstrated that the administration of BM ameliorated memory deficits and protected the neuronal cells against brain damage.1–6) Evidence indicates that BM reduces oxidative stress in the brain and thereby protecting neuronal cells from oxidative stress-induced cytotoxicity.7,8) It also reduces not only hippocampal β-amyloid deposition but also stress-induced hippocampal damage.9)
We previously demonstrated that BM extract and the cholinesterase inhibitor tacrine attenuated olfactory bulbectomy (OBX)-induced cognitive deficits and also that these effects were, at least partly, mediated by enhancement of synaptic plasticity-related signaling and brain-derived neurotrophic factor (BDNF) transcription as well as by the protection of cholinergic systems from OBX-induced neuronal damage. Our findings suggested that treatments with BM are beneficial for dementia and other neurodegenerative disorders.10) This idea was further supported by our previous studies using in vivo and in vitro models of cerebral ischemia. In an in vivo study using a transient two vessel occlusion mouse model (T2VO), we found that the standardized BME containing 21.8% bacoside A and 11.0% bacopaside I attenuated transient cerebral ischemia-induced cognitive deficits. Moreover, our in vitro study using organotypic hippocampal slice cultures (OHSCs) revealed that bacopaside I, a major triterpenoid from BM, protected against oxygen- and glucose-deprivation (OGD)-induced hippocampal cell damage in via protein kinase C (PKC) and phosphatidylinositol 3-kinase (PI3K)/Akt mechanisms.11) However, it currently remains unclear whether the anti-dementia effects of BM extract involves endogenous neurogenesis.
Trimethyltin (TMT) is an organotin compound with potent neurotoxicant effects that induces neuronal degeneration in human and rodent central nervous systems.12) According to previous findings,13–15) a single treatment with TMT caused neuronal cell loss in restricted brain regions, such as the dentate gyrus, olfactory bulb, anterior olfactory nucleus, and frontal cerebral cortex, in mice. Moreover, a treatment with TMT markedly enhanced neurogenesis in the dentate gyrus and olfactory bulb by inducing the proliferation of the endogenous neural stem/progenitor cells (NPCs) in each of these brain regions.16,17) In rodents, TMT was found to cause hippocampus-dependent cognitive deficits, including short-term memory, as measured in the passive avoidance and novel object recognition testes18–21); spatial working memory using the modified Y- and radial arm mazes21,22); and spatial long-term memory using the Morris water maze.21,23,24) Furthermore, behavioral studies demonstrated that TMT induced abnormal behaviors such as locomotor activity or depressive-like behaviors.17,21,25) Therefore, TMT-intoxicated rodents are a useful model of degenerative diseases, such as Alzheimer’s disease, the most common cause of dementia.26)
The present study aimed to clarify the effects of BM on neuronal repair following hippocampal neuronal damage by using a mouse model of TMT-induced neuronal loss/self-repair in the hippocampal dentate gyrus. The results obtained demonstrated that BME ameliorated cognitive impairments and enhanced neuroregeneration in the TMT-treated mice.
MATERIALS AND METHODS
Preparation of Bacopa monnier ExtractThe plant Bacopa monnier (L.) WETTST. (BM) used in the present study was collected in Ho Chi Minh city, Vietnam and identified by Dr. Pham Thanh Huyen of the Department of Resource Medicinal Material, National Institute of Medicinal Material, Hanoi, Vietnam (NIMM) and stored as voucher specimen 9967. The BME extract used in this study was the same as that used in the previous study.11) Briefly, the aerial part of the plant was collected, dried in a hot-air oven at 50°C, cut into small pieces and then crushed. The herbal powder was extracted with 50% ethanol (1 : 8 w/v) at 85°C for 2 h. This extraction process was repeated. The extracts were combined, filtrated, and then concentrated under reduced pressure at 50°C. Further extraction was conducted 3 times using n-butanol. The yield of the extraction from the dried herb was calculated as 8.3% (w/w). The extract was subjected to high-performance liquid chromatography as described in our previous paper.11) Dried BM extract was estimated to contain 21.8% bacoside A and 11.0% bacopaside.
Animals and Drug TreatmentMale Swiss albino mice (National Institute of Hygiene and Epidemiology, Hanoi, Vietnam) and male ddY mice (Japan, SLC, Shizuoka, Japan) were obtained at the age of 4 weeks old. The animals were habituated to the laboratory animal room at least for one week before the start of the experiments. Animal were given food and water ad libitum. Housing was thermostatically maintained at 24 ± 1°C with 65 ± 5% humidity and 12-h light–dark cycle (lights on: 07:00–19:00). Behavioral experiments were performed during the light phase between 9:00 and 18:00 according to the experimental schedules described in Fig. 1. The present study was conducted in accordance with the Committee for the Ethical Use of Experimental Animals, Setsunan University.
Animals were intraperitoneally (i.p.) injected with TMT (2.8 mg/kg) dissolved in phosphate-buffered saline (PBS) to prepare a mouse model of neuronal loss in the hippocampal dentate gyrus (impaired mice). Control animals received vehicle PBS (naïve mice). TMT-treated animals were further divided into two groups: a TMT control group (TMT-control) and a TMT-BME group (TMT-BME). The TMT control and TMT-BME groups per orally administered distilled water and BME (50 mg/kg), respectively, once daily from day 2 after the TMT treatment during the experimental period (Fig. 1). BME was dissolved in water immediately before its administration. To elucidate the effects of BME on mitotic cells in TMT-treated animals, animals were administered BrdU (50 mg/kg, i.p.) twice in a 12-h interval on day 2 after the TMT treatment (Fig. 1B). They were decapitated on days 17 or 32 after the TMT treatment to prepare hippocampal slices for histochemistry.
Behavioral StudyObject Location TestThe object location test (OLT) was performed 15 and 30 d after the BME treatment according to the method described by Zhao et al.27) Briefly, animals were individually acclimatized to an observation box (35 × 35 × 50 cm) for 10 min on 1 d before the experiment. The OLT consisted of sample and test phase trials. In the sample phase trial, each mouse was placed in the observation box for 5 min and exposed to two identical objects, objects 1 and 2, which were separately placed on the arena. The total time spent by a mouse to explore each object was measured, and the mouse was then returned to the home cage. The test phase trial was conducted 10 min after the sample phase trial. In this trial, objects were replaced by their identical copies, one of which was placed in the same position, whereas the other was moved to the adjacent corner so that the two objects were in diagonally opposite corners. Mice were exposed to the objects for 5 min and the total time spent exploring each object was measured.
Modified Y-Maze TestThe modified Y-maze test was conducted according to the method described by Yamada et al.28) on days 18 and 33 after the initiation of the BME treatment. The apparatus used for this test consisted of black polypropylene walls with 3 arms that were each 40 cm long, 12 cm wide at the top, 3 cm wide at the bottom, and 18 cm high. This test was a two-trial task with a sample trial and a test trial that were separated by an inter-trial interval. In the sample test, each mouse was individually placed in the maze with one of the 3 arms closed. The animal was allowed to explore the other 2 arms freely for 5 min. In the test trial conducted 30 min after the sample trial, the animal was again placed in the maze with all 3 arms opened, and allowed to explore the arms freely. The previously closed arm that was opened in the test trial was defined as the novel arm. Animal behavior was video-recorded for analysis using Any Maze system® (Stoelting Co., IL, U.S.A.).
Passive Avoidance TestA passive avoidance test was conducted using a step-through paradigm. The apparatus used in the test consisted of light and dark compartments separated by an automatic guillotine door. Each compartment had a grid floor through which electric footshocks were delivered. The test consisted of training and test trials. In the training trials, mice were individually placed into the light compartment, facing away from the dark compartment. Animals were allowed to explore freely for 10 s and then the guillotine door was lifted. When all four paws of the animal entered the dark compartment, the guillotine door was closed. Three sec after the door was closed, animals received electric footshocks (0.5 mA, 3 s duration) through the grid floor for 10 s and were then returned to their home cages. Test trials were conducted 24 h after training. In the test trials, the mouse was returned to the lighted compartment, facing away from the dark compartment. After 10 s, the guillotine door was lifted and latency until all four paws of the animal entered the dark compartment was recorded as an index of fear-based learning and memory performance.
Neurochemical StudyNissl Staining of Hippocampal TissuesOn day 17 after the treatment with TMT, mice were anesthetized with mixed anesthesia (0.75 mg/kg medetomidine, 4 mg/kg midazolam, and 5 mg/kg butorphanol tartrate) and were perfused intracardially with a heparinized saline followed by 10% formalin saline. The brains were excised and post-fixed for 3 h in saline containing 10% formalin. The brains then were immersed in a graded strength of sucrose solutions (12, 15 and 18%) for 48 h. Coronal sections (20 µm thick) were taken at the approximate level of the anterior hippocampus (Bregma −2.06 to −2.30 mm) using a freezing microtome and stained with 0.5% cresyl violet. Three sagittal sections prepared from the brain of each mouse were used for Nissl staining. The images of CA1 and CA3 regions was captured using microscope (Olympus PROVIS®, Olympus Corporation, Tokyo, Japan) and the average intensity was analyzed by Image-J software (ver. 1.41, NIH; Bethesda, MD, U.S.A.).
Elucidation of Neuroprotective Effects of BME on TMT-Induced Excitotoxicity Model of Organotypic Hippocampal Slice Cultures (OHSCs)OHSCs were prepared from 7-d-old ICR mice as previously described29,30) and cultivated for 8 d before use. In experiments, five OHSCs from the brain of each mouse were incubated with test drugs for 30 min and then treated with TMT in the presence of the test drugs for 24 h. After washing out the drugs, OHSCs were further incubated in normal medium for 24 h. TMT-induced hippocampal damage was quantified by densitometric measurement of the cellular uptake of propidium iodide (PI) (Sigma, St. Louis, MO, U.S.A.) as previously described.29,30) PI-fluorescence (630 nm) images were captured using a confocal laser microscope (TCS-SP5, Leica Microsystems, Tokyo, Japan) and the average intensity was analyzed by Image-J software (ver. 1.41, NIH). The value obtained from control OHSCs exposed to 10 µM TMT was set to 100% damage and was then compared with the values obtained from OHSCs treated with the test drugs.
ImmunohistochemistryImmunohistochemical studies using the hippocampus were conducted according to the method described by Yoneyama et al.31) 17 and 32 d after starting the admnistration of BME (50 mg/kg). In brief, mice were anesthetized with chloral hydrate (500 mg/kg, i.p.) and fixed by intracardic perfusion with PBS followed by 4% (w/v) paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4). The brains were quickly removed and further fixed with the same fixative solution at 4°C overnight. Post-fixed brains were embedded in paraffin, cut with a microtome into seven sagittal sections with a thickness of 3–5 µm thickness at 100 µm intervals in a range between 0.9 and 1.6 mm relative to the lateral, according to the atlas of Paxinos and Franklin,32) and then placed on Matsunami adhesive saline-coated glass slides (Matsunami Glass, Kyoto, Japan). The paraffin-embedded brain sections were then deparaffinized with xylene, rehydrated by immersion in ethanol of graded decreasing concentrations of 100 to 50% (v/v), and finally washed with water. The sections obtained were subjected to the immunohistochemical procedures. Sections were initially heated in 10 mM sodium citrate buffer at pH 7.0 for 10 min in a microwave oven then washed with Tris-buffered saline with Tween 20 (PBS-T). The slices were then blocked with 5% normal goat serum at room temperature for 1 h and incubated with the primary antibody against BrdU (3 µg/mL) at 4°C overnight. After washing with PBS-T, they were incubated with the secondary antibody (Alexa-Fluor 594-conjugated anti-rat IgG antibody for BrdU) at room temperature for 2 h. Sections were viewed with a BX41 microscope (Olympus) equipped with a DS-Ri1 camera (Nikon, Tokyo, Japan), and the number of strongly labeled cells was counted via microscopic observations. To obtain the total number of BrdU-positive cells per animal, we used seven sagittal sections prepared from the brain of each mouse for immunostaining and counted BrdU-positive cells in the whole dentate gyrus (granule cell layer-GCL, subgranular zone-SGZ, hilus, molecular layer) and GCL ± SGZ.
Statistical AnalysisData were expressed as the mean ± standard error of the mean (S.E.M.). All of the data obtained in the present study, except those from the OLT, were analyzed by a one-way ANOVA followed by a post hoc comparison test (Student–Newman–Keuls/Dunnett). OLT data were analyzed using a paired Student’s t-test. Differences of p < 0.05 were considered to be significant.
RESULTS
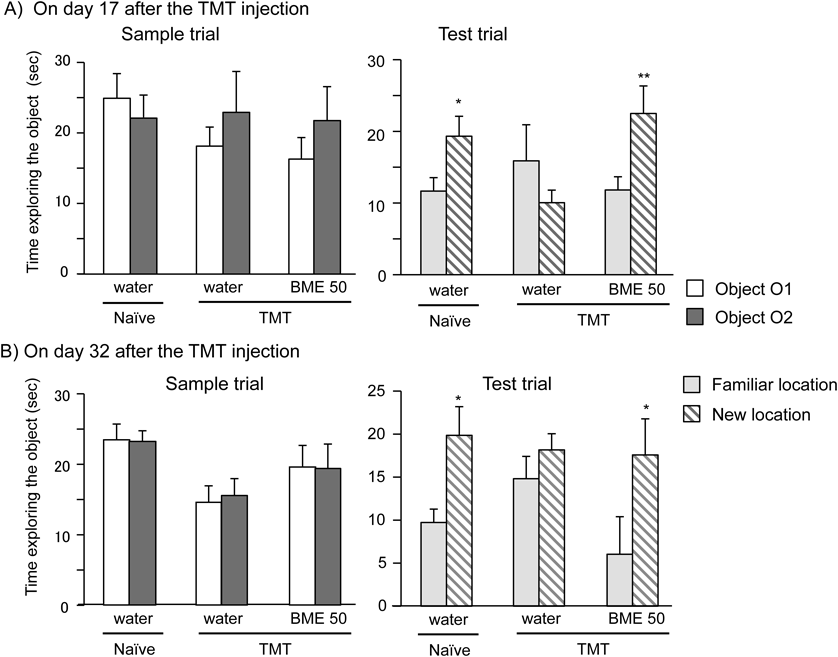
Effects of BME on Spatial Working Memory Deficits Caused by TMTThe spatial working memories of mice were elucidated twice using the OLT and modified Y-maze test, which were conducted on days 17–20 and 32–35, respectively, in a 15-d interval. In the sample trials of the OLT on days 17 and 32, animals that had been administered the test drugs or vehicle daily prior the test showed no significant difference in preference to the same objects and spent almost the same time exploring each object. However, in the test trials in which one of the objects was placed in a new location and the other in a familiar location, naïve control animals spent significantly more time exploring the object placed in the new location than that placed in the familiar location. This spatial working memory is relevant to episodic memory in humans. As shown in Fig. 2, the TMT-treated group failed to discriminate between two objects placed in familiar and novel locations in the OLT on days 17 and 32 after the TMT treatment, indicating that animals treated with TMT exhibited deficits in spatial working memory. However, when the TMT group was treated with BME for 15 and 30 d prior to the behavioral experiments, they were able to recognize a difference in the objects placed in the novel and familiar locations and spent significantly more time exploring the object placed in the novel location (after 15 d of the BME treatment: naïve-control: p < 0.05, BME-treated TMT: p < 0.01, Wilcoxon signed rank test; after 30 d of the BME treatment: naïve-control: t = −2.319, df = 9; p < 0.05; BME-treated TMT: t = −2.573, df = 9; p < 0.05, paired t-test) (Fig. 2).
To further confirm the effects of TMT and BME on spatial working memory in mice, we conducted the modified Y-maze test on days 20 and 35 using the same animals (Figs. 3A, B). In the test trials, we measured the time the animal spent exploring each arm during a 5-min observation period. As shown in Fig. 3B, the % time the naïve control animal spent in a new arm was more than a chance level of 33%. This index of the % time spent in a new arm represents spatial working memory. The % time of the vehicle-treated TMT group was significantly less than that of the naïve control animals, indicating TMT-induced impairments in spatial working memory. However, the administration of BME for 18 and 33 d significantly prolonged the time the TMT-treated animals spent exploring the new arms. (after 18 d of the BME treatment: [F(2, 28) = 3.963, p = 0.031; water-treated TMT group vs. naïve group, p = 0.030; water-treated TMT group vs. BME-treated TMT group, p = 0.049]; after 33 d of the BME treatment: [F(2, 26) = 5.027, p = 0.014; water-treated TMT group vs. naïve group, p = 0.017; water-treated TMT group vs. BME-treated TMT group, p = 0.018] (Fig. 3).
Effects of BME on the Fear-Associated Cognitive Performance of TMT-Treated Mice in the Passive Avoidance TestWe also examined the effects of BME on TMT-induced deficits in fear-associated cognitive performance using the passive avoidance test (Fig. 4). Vehicle-treated mice showed a significant decrease in latency (approximately 61.5%, [F(2, 15) = 11.626, p < 0.001; water-treated TMT group vs. naïve group, p = 0.001]) to enter the dark compartment of the passive avoidance apparatus from that of the naïve control. On the other hand, the daily administration of BME (50 mg/kg) for 30 d significantly reversed the memory deficit induced by TMT [F(2, 15) = 11.626, p = 0.001; water-treated TMT group vs. BME-treated TMT group, p = 0.008].
BME Exerts Neuroprotective Effects on TMT-Induced Hippocampal Cell Damage in Vivo and in VitroBased on the present results that treatment with BME protected the spatial cognitive function of mice against TMT-induced neurotoxicity, we elucidate the effects of BME on hippocampal cells after TMT treatment in vivo and in vitro. As shown in Fig. 5, the cresyl violet staining of hippocampal tissues revealed that the number of pyramidal cells in the hippocampal CA1 and CA3 regions was significantly reduced 17 d after the TMT treatment and also that the administration of BME prevented the TMT-induced damage in the CA1 and CA3 regions.
The neuroprotective effects of BME on TMT-induced hippocampal cell damage were also observed in the present in vitro study using OHSCs. An in vitro treatment with TMT dose-dependently induced hippocampal cell damages detected by PI uptake in OHSCs. It has been reported that TMT causes endogenous glutamate release from hippocampal slice, so it linked to excitotoxicity.33) In this study, MK-801, an N-methyl-D-aspartate receptor antagonist, was employed as a positive control in TMT-induced neurotoxicity in OHSCs. Our results showed that MK-801 (30 mM) significantly reduces TMT-induced cell damages in hippocampal slice culture. Interestingly, the damage caused by 10 µM TMT was significantly attenuated by BME (20 and 30 µg/mL) (Fig. 6).
BME Increased the Number of BrdU-Incorporating Cells Generated Following Neuronal Loss in the Dentate Gyrus Mice of TMT-Treated MiceTMT reportedly induces neuronal loss/self-repair in the hippocampal dentate gyrus region. As shown in Fig. 5, the TMT treatment markedly increased the number of BrdU-immunopositive cells in the granular cell layer (GCL) and subgranular zone of the dentate gyrus as well as the total number of BrdU-positive cells in the whole dentate gyrus region on day 15 (in the GCL ± SGZ: [F(2, 9) = 6.160, p = 0.021; water-treated TMT group vs. naïve group, p = 0.027; in the whole dentate gyrus: [F(2, 9) = 6.268, p = 0.02; water-treated TMT group vs. naïve group, p = 0.015) and day 30 (in the GCL ± SGZ: [F(2, 9) = 17.218, p < 0.001; water-treated TMT group vs. naïve group, p = 0.025; in the whole dentate gyrus: F(2, 9) = 17.218, p < 0.001; water-treated TMT group vs. naïve group, p = 0.001) after starting treatment with water or BME (Fig. 7). The daily administration of BME (50 mg/kg) for 15 d had no effect on the TMT-induced increase in BrdU-immunopositive cells in the dentate gyrus; however, when BME was given for 30 d, it significantly enhanced the neurogenerative effects of TMT on the dentate gyrus region (in the GCL ± SGZ: [F(2, 9) = 17.218, p < 0.001; water-treated TMT group vs. BME-treated TMT group, p = 0.011]; in the whole dentate gyrus: [F(2, 9) = 57.502, p < 0.001; water-treated TMT group vs. BME-treated TMT group, p < 0.001).
DISCUSSION
In the present study using a TMT model of neuronal loss/self-repair, we demonstrated that BME attenuated TMT-induced cognitive deficits in mice and potentiated TMT-induced neurogenesis following neuronal loss in the hippocampal dentate gyrus. Our results suggest that BME exerted ameliorative effects on TMT-induced cognitive deficits by suppressing neurodegeneration and facilitating neuro-regeneration in the hippocampus of TMT-treated mice.
Previous studies demonstrated that TMT induced behavioral and cognitive deficits in humans and rodents and also that TMT-induced cognitive dysfunction was due to neurodegeneration that occurred within several days of the TMT treatment in hippocampal sub-regions, such as CA1 and dentate gyrus areas.34,35) Moreover, these hippocampal sub-regions, which are susceptible to TMT, have been shown to plays an important role in spatial working and reference memories. In the present study, we evaluated the spatial cognitive performance of TMT-treated animals using the OLT and modified Y-maze test. The results obtained revealed that the daily administration of BME attenuated spatial working memory deficits induced by the TMT treatment. Thus, when taken together with previous findings, the present results indicate that BME protects or rescues neurons, particularly in the hippocampal CA1 regions, from TMT-induced neurodegeneration. Indeed, this idea was supported by the present study using Nissl staining that TMT-induced neuronal cell lose in the CA1 and CA3 regions was attenuated in animals treated with daily administration of BME for 17 d.
The present study also revealed that the daily administration of BME attenuated TMT-induced impairments in learning and memory performance in the passive avoidance test conducted 35 d after the TMT treatment. This result is consistent with previous findings,19,23) indicating that the TMT treatment induces long-lasting cognitive dysfunction which is attributable to neuronal lesions caused not only in the hippocampus, but also the amygdala because the amygdala also plays an important role in fear-aggravated cognitive performance.36,37) Moreover, TMT reportedly causes neurodegeneration in the hippocampus and other brain regions, which become evident several days after treatment; however, there have been some conflicting findings.17,38,39) Therefore, the administration of BME after a treatment with TMT may exert a preventive/rescuing effect on the neuronal damages induced in the early stage after the TMT treatment.
The present results provide further support to our idea that BME is beneficial for the treatment of cognitive dysfunctions caused by various insults related to cerebral ischemia and Alzheimer’s disease, which lead to neuronal damage/degeneration.10,11) We previously demonstrated that BME ameliorated OBX-induced cognitive impairments in mice and that this effect was attributable to several mechanisms, such as the inhibition of acetylcholinesterase activity, enhanced synaptic plasticity-related signaling in the hippocampus, and the prevention of OBX-induced medial septal cholinergic neurons projecting to the hippocampus.10) On the other hand, evidence indicates that TMT-induced cognitive deficits are also mediated by dysfunctional central cholinergic systems23) as well as neurosignaling systems including BDNF and cAMP response element-binding protein (CREB) in the hippocampus.24,40) Taken together with these findings, the present results lead us to suggest that BME attenuated TMT-induced cognitive impairments in mice, at least in part by protecting the cholinergic system.
Neurogenesis was markedly enhanced in all sub-regions of the dentate gyrus on days 17 and 32 after TMT and that the administration of BME for 30 d, but not 15 d further promoted TMT-induced enhancements in neurogenesis in these regions. The hippocampal dentate gyrus in adult animals is known to contain neural progenitors that proliferate and differentiate into neurons in response to brain injury.41) Moreover, previous studies reported that TMT induced neurodegeneration in the hippocampal CA1 and CA3 regions42) in an early stage (2–5 d) after the TMT treatment, whereas it enhanced neurogenesis in the dentate gyrus and olfactory bulb in a late stage (>15 d) after the TMT treatment through the proliferation of endogenous NPCs in these regions.17,31) The molecular mechanisms involved in TMT-induced neurogenesis in the dentate gyrus region and how BME potentiates the effects of TMT in that region in a late stage after a TMT treatment currently remain unclear. However, some explanations have been proposed. BME may exert enhancing effects on TMT-induced neurogenesis in dentate gyrus regions by increasing the survival rate of differentiated neuronal cells in that region. In our previous study using organotypic hippocampal slice cultures, we found that, among bacopasides, which are major constituents of BME, bacopaside I not only suppressed the OGD-induced apoptosis of neuronal cells, but also protected neuronal cells via activation of PKC and PI3/Akt mechanisms.11) Furthermore, a long-term treatment with BME may enhance neurogenesis in dentate gyrus regions because the activation of an Akt-mediated signaling pathway have been reported in adult hippocampal neurogenesis.43) Further studies are required to elucidate the neuroprotective and neurogenerative effects of BME in the hippocampus. Since the present results showed that the BME treatment for 15 d did not affect TMT-induced neurogenesis in the hippocampus, the ameliorative effects of this plant extract on cognitive dysfunction observed 17 d after the TMT do not appear to be due to apparent BME-induced enhancement in neurogenesis.
In conclusion, the present study demonstrated that TMT causes cognitive deficits relevant to neuronal degeneration in the CA1 area and neuroregeneration in the dentate gyrus region and also that the daily administration of BME ameliorated TMT-induced cognitive disturbances mainly by suppressing TMT-induced neurodegeneration in the hippocampus.
Acknowledgments
The present work was supported in part by a Grant from National Institute of Medicinal Materials (NIMM) and FIRST project (07/FIRST/1.a/NIMM).
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Brown WR, Thore CR. Review: cerebral microvascular pathology in ageing and neurodegeneration. Neuropathol. Appl. Neurobiol., 37, 56–74 (2011).
- 2) Kishore K, Singh M. Effect of bacosides, alcoholic extract of Bacopa monniera Linn. (brahmi), on experimental amnesia in mice. Indian J. Exp. Biol., 43, 640–645 (2005).
- 3) Kumar SS, Saraswathi P, Vijayaraghavan R. Effect of bacopa monniera on cold stress induced neurodegeneration in hippocampus of wistar rats: a histomorphometric study. J. Clin. Diagn. Res., 9, AF05–AF07 (2015).
- 4) Saini N, Singh D, Sandhir R. Neuroprotective effects of Bacopa monnieri in experimental model of dementia. Neurochem. Res., 37, 1928–1937 (2012).
- 5) Saraf MK, Prabhakar S, Anand A. Neuroprotective effect of Bacopa monniera on ischemia induced brain injury. Pharmacol. Biochem. Behav., 97, 192–197 (2010).
- 6) Vohora D, Pal SN, Pillai KK. Protection from phenytoin-induced cognitive deficit by Bacopa monniera, a reputed Indian nootropic plant. J. Ethnopharmacol., 71, 383–390 (2000).
- 7) Simpson T, Pase M, Stough C. Bacopa monnieri as an antioxidant therapy to reduce oxidative stress in the aging brain. Evid. Based Complement. Alternat. Med., 2015, 615384 (2015).
- 8) Dhanasekaran M, Tharakan B, Holcomb LA, Hitt AR, Young KA, Manyam BV. Neuroprotective mechanisms of ayurvedic antidementia botanical Bacopa monniera. Phytother. Res., 21, 965–969 (2007).
- 9) Chaudhari KS, Tiwari NR, Tiwari RR, Sharma RS. Neurocognitive effect of nootropic drug brahmi (Bacopa monnieri) in Alzheimer’s disease. Ann. Neurosci., 24, 111–122 (2017).
- 10) Le XT, Pham HT, Do PT, Fujiwara H, Tanaka K, Li F, Van Nguyen T, Nguyen KM, Matsumoto K. Bacopa monnieri ameliorates memory deficits in olfactory bulbectomized mice: possible involvement of glutamatergic and cholinergic systems. Neurochem. Res., 38, 2201–2215 (2013).
- 11) Le XT, Nguyet Pham HT, Van Nguyen T, Minh Nguyen K, Tanaka K, Fujiwara H, Matsumoto K. Protective effects of Bacopa monnieri on ischemia-induced cognitive deficits in mice: the possible contribution of bacopaside I and underlying mechanism. J. Ethnopharmacol., 164, 37–45 (2015).
- 12) Fiedorowicz A, Figiel I, Kaminska B, Zaremba M, Wilk S, Oderfeld-Nowak B. Dentate granule neuron apoptosis and glia activation in murine hippocampus induced by trimethyltin exposure. Brain Res., 912, 116–127 (2001).
- 13) Ogita K, Nitta Y, Watanabe M, Nakatani Y, Nishiyama N, Sugiyama C, Yoneda Y. In vivo activation of c-Jun N-terminal kinase signaling cascade prior to granule cell death induced by trimethyltin in the dentate gyrus of mice. Neuropharmacology, 47, 619–630 (2004).
- 14) Kawada K, Yoneyama M, Nagashima R, Ogita K. In vivo acute treatment with trimethyltin chloride causes neuronal degeneration in the murine olfactory bulb and anterior olfactory nucleus by different cascades in each region. J. Neurosci. Res., 86, 1635–1646 (2008).
- 15) Shuto M, Seko K, Kuramoto N, Sugiyama C, Kawada K, Yoneyama M, Nagashima R, Ogita K. Activation of c-Jun N-terminal kinase cascades is involved in part of the neuronal degeneration induced by trimethyltin in cortical neurons of mice. J. Pharmacol. Sci., 109, 60–70 (2009).
- 16) Yoneyama M, Kawada K, Ogita K. Enhanced neurogenesis in the olfactory bulb in adult mice after injury induced by acute treatment with trimethyltin. J. Neurosci. Res., 88, 1242–1251 (2010).
- 17) Ogita K, Nishiyama N, Sugiyama C, Higuchi K, Yoneyama M, Yoneda Y. Regeneration of granule neurons after lesioning of hippocampal dentate gyrus: evaluation using adult mice treated with trimethyltin chloride as a model. J. Neurosci. Res., 82, 609–621 (2005).
- 18) Onaka Y, Wada S, Yamaguchi T, Yoneyama M, Ogita K. Preventive effect of olanzapine on trimethyltin neurotoxicity in mice: evaluation of hippocampal neuronal loss, microglial activation, and cognitive dysfunction. Global Drugs and Therapeutics, 3, 1–5 (2018).
- 19) Kim CR, Choi SJ, Kwon YK, Kim JK, Kim YJ, Park GG, Shin DH. Cinnamomum loureirii extract inhibits acetylcholinesterase activity and ameliorates trimethyltin-induced cognitive dysfunction in mice. Biol. Pharm. Bull., 39, 1130–1136 (2016).
- 20) Walsh TJ, Gallagher M, Bostock E, Dyer RS. Trimethyltin impairs retention of a passive avoidance task. Neurobehav. Toxicol. Teratol., 4, 163–167 (1982).
- 21) Kim JM, Park SK, Kang JY, Park SB, Yoo SK, Han HJ, Kim CW, Lee U, Kim SH, Heo HJ. Ethyl acetate fraction from Persimmon (Diospyros kaki) ameliorates cerebral neuronal loss and cognitive deficit via the JNK/Akt pathway in TMT-induced mice. Int. J. Mol. Sci., 19, 1499 (2018).
- 22) Walsh TJ, Miller DB, Dyer RS. Trimethyltin, a selective limbic system neurotoxicant, impairs radial-arm maze performance. Neurobehav. Toxicol. Teratol., 4, 177–183 (1982).
- 23) O’Connell A, Earley B, Leonard BE. The neuroprotective effect of tacrine on trimethyltin induced memory and muscarinic receptor dysfunction in the rat. Neurochem. Int., 25, 555–566 (1994).
- 24) Park HJ, Shim HS, Ahn YH, Kim KS, Park KJ, Choi WK, Ha HC, Kang JI, Kim TS, Yeo IH, Kim JS, Shim I. Tremella fuciformis enhances the neurite outgrowth of PC12 cells and restores trimethyltin-induced impairment of memory in rats via activation of CREB transcription and cholinergic systems. Behav. Brain Res., 229, 82–90 (2012).
- 25) Yoneyama M, Shiba T, Hasebe S, Umeda K, Yamaguchi T, Ogita K. Lithium promotes neuronal repair and ameliorates depression-like behavior following trimethyltin-induced neuronal loss in the dentate gyrus. PLOS ONE, 9, e87953 (2014).
- 26) Woodruff ML, Baisden RH. Trimethyltin neurotoxicity in the rat as an analogous model of Alzheimer’s disease. Springer Science+Business Medica, LLC, New York (1994).
- 27) Zhao Q, Murakami Y, Tohda M, Obi R, Shimada Y, Matsumoto K. Chotosan, a kampo formula, ameliorates chronic cerebral hypoperfusion-induced deficits in object recognition behaviors and central cholinergic systems in mice. J. Pharmacol. Sci., 103, 360–373 (2007).
- 28) Yamada M, Hayashida M, Zhao Q, Shibahara N, Tanaka K, Miyata T, Matsumoto K. Ameliorative effects of yokukansan on learning and memory deficits in olfactory bulbectomized mice. J. Ethnopharmacol., 135, 737–746 (2011).
- 29) Inada C, Niu Y, Matsumoto K, Le TX, Fujiwara H. Possible involvement of VEGF signaling system in rescuing effect of endogenous acetylcholine on NMDA-induced long-lasting hippocampal cell damage in organic hippocampal slice cultures. Neurochem. Int., 75, 39–47 (2014).
- 30) Inada C, Thi Le X, Tsuneyama K, Fujiwara H, Miyata T, Matsumoto K. Endogenous acetylcholine rescues NMDA-induced long-lasting hippocampal cell damage via stimulation of muscarinic M(1) receptors: elucidation using organic hippocampal slice cultures. Eur. J. Pharmacol., 699, 150–159 (2013).
- 31) Yoneyama M, Tanaka M, Hasebe S, Yamaguchi T, Shiba T, Ogita K. Beneficial effect of cilostazol-mediated neuronal repair following trimethyltin-induced neuronal loss in the dentate gyrus. J. Neurosci. Res., 93, 56–66 (2015).
- 32) Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. Academic Press, San Diego (2004).
- 33) Patel M, Ardelt BK, Yim GK, Isom GE. Interaction of trimethyltin with hippocampal glutamate. Neurotoxicology, 11, 601–608 (1990).
- 34) Ceccariglia S, D’Altocolle A, Del Fa A, Pizzolante F, Caccia E, Michetti F, Gangitano C. Cathepsin D plays a crucial role in the trimethyltin-induced hippocampal neurodegeneration process. Neuroscience, 174, 160–170 (2011).
- 35) Geloso MC, Corvino V, Michetti F. Trimethyltin-induced hippocampal degeneration as a tool to investigate neurodegenerative processes. Neurochem. Int., 58, 729–738 (2011).
- 36) Park HJ, Lee SY, Shim HS, Kim JS, Kim KS, Shim I. Chronic treatment with Squid phosphatidylserine activates glucose uptake and ameliorates TMT-induced cognitive deficit in rats via activation of cholinergic systems. Evid. Based Complement. Alternat. Med., 2012, 601018 (2012).
- 37) Whittington DL, Woodruff ML, Baisden RH. The time–course of trimethyltin-induced fiber and terminal degeneration in hippocampus. Neurotoxicol. Teratol., 11, 21–33 (1989).
- 38) Shuto M, Higuchi K, Sugiyama C, Yoneyama M, Kuramoto N, Nagashima R, Kawada K, Ogita K. Endogenous and exogenous glucocorticoids prevent trimethyltin from causing neuronal degeneration of the mouse brain in vivo: involvement of oxidative stress pathways. J. Pharmacol. Sci., 110, 424–436 (2009).
- 39) Yoneyama M, Hasebe S, Kawamoto N, Shiba T, Yamaguchi T, Kikuta M, Shuto M, Ogita K. Beneficial in vivo effect of aripiprazole on neuronal regeneration following neuronal loss in the dentate gyrus: evaluation using a mouse model of trimethyltin-induced neuronal loss/self-repair in the dentate gyrus. J. Pharmacol. Sci., 124, 99–111 (2014).
- 40) Park HR, Lee H, Park H, Cho WK, Ma JY. Fermented Sipjeondaebo-tang alleviates memory deficits and loss of hippocampal neurogenesis in scopolamine-induced amnesia in mice. Sci. Rep., 6, 22405 (2016).
- 41) Taupin P, Gage FH. Adult neurogenesis and neural stem cells of the central nervous system in mammals. J. Neurosci. Res., 69, 745–749 (2002).
- 42) Cannon RL, Hoover DB, Baisden RH, Woodruff ML. The effect of time following exposure to trimethyltin (TMT) on cholinergic muscarinic receptor binding in rat hippocampus. Mol. Chem. Neuropathol., 23, 47–62 (1994).
- 43) Garza JC, Guo M, Zhang W, Lu XY. Leptin increases adult hippocampal neurogenesis in vivo and in vitro. J. Biol. Chem., 283, 18238–18247 (2008).