Abstract
Androgen receptor (AR) has a key role in the development and progression of prostate cancer, and AR antagonists are used for its remedy. Recently, carborane derivatives, which are carbon-containing boron clusters have attracted attention as new AR ligands. Here we determined the force field parameters of 10-vertex and 12-vertex p-carborane to facilitate in silico drug design of boron clusters. Then, molecular dynamics (MD) simulations of complexes of AR-carborane derivatives were performed to evaluate the parameters and investigate the influences of carborane derivatives on the three-dimensional structure of AR. Energy profiles were obtained using quantum chemical calculations, and the force-field parameters were determined by curve fitting of the energy profiles. The results of MD simulations indicated that binding of the antagonist–BA341 changed some hydrogen-bond formations involved in the structure and location of helix 12. Those results were consistent with previously reported data. The determined parameters are also useful for refining the structure of the carborane-receptor complex obtained by docking simulations and development of new ligands with carborane cages not only for AR but also for various receptors.
INTRODUCTION
The exceptional hydrophobic character and spherical geometry of dicarba-closo-dodecaboranes (12-vertex carboranes), which is one of the carbon-containing boron clusters, have been reported1–5) (Fig. 1A). To develop new ligands, hydrophobicity is an important factor because ligand-binding pockets can be detected by hydrophobic residues in proteins.6,7) Because the electronegativities of boron and hydrogen are nearly equal, the hydrophobicity of carboranes is expected to be appropriate for the hydrophobic building block of ligands. Since the structure of carboranes are extremely rigid, entropy loss upon docking into ligand-binding pockets is minimal. Furthermore, compared to hydrocarbons, such as adamantine, with many carbon atoms that must be distinguished to eliminate isomeric side products, the introduction of functional groups as substituent to the specific position of carborane is easier.
We have focused on the androgen receptor (AR) as the target for carboran derivatives. AR is a member of the nuclear receptor transcriptional superfamily, which mediates several gene expressions through androgen binding.8,9) AR consists of an N-terminal domain (NTD), DNA-binding domain (DBD), flexible and short hinge region, and a C-terminal ligand-binding domain (LBD), which contains the second activation function site (AF-2). The AF-2, which is composed of helices 3, 4, 5, and 12 (H3, H4, H5, and H12), activates the recruitment of coactivator and facilitates transcription in a ligand-dependent manner.10,11) In the ligand-free state, the transcriptional activity of AR is inhibited by the corepressor, which interacts with the DBD; in contrast, agonist binding causes conformational change of H12 close to the ligand-binding pocket and corepressor dissociation.9,12,13) Instead, the coactivator binds to NTD, DBD or LBD and enhances transcriptional activity of AR. In the antagonist form, H12 is positioned in a different orientation, thereby opening the entrance to the LBD in comparison with that in its agonist form, and this conformational change is believed to be sufficient for inhibiting coactivator binding. The androgen activity is important in prostate maintenance and plays a key role in the development and progression of prostate cancer; therefore, AR antagonists are used as a remedy for prostate cancer.10,14,15) However, the survival and QOL in prostate-cancer patients have not been sufficiently improved by existing therapies.15–17) In addition, AR antagonists are also considered to be effective against AR-positive breast cancer.18) Therefore, inventions of novel ligands for AR are required for these therapies.
In previous studies, we have revealed that some carborane derivatives are ligands for AR and estrogen receptors.19,20) One of the 12-vertex para-carborane derivatives, BA341, particularly indicates higher AR-binding affinity and antagonist activity than those of a known AR antagonist–hydroxyflutamide, whereas the result of docking simulation has suggested that the binding mode of BA341 is similar to that of an AR agonist, dehydroteststeron (DHT).21) The docking result also indicates that the activities of carborane derivatives depend on the high hydrophobicity and bulkiness of the boron-rich cluster. In fact, hexadecahedral 1,10-dicarba-closo-decaborane, which is generally called 10-vertex para-carborane (Fig. 1B), is known as a partial agonist for AR.22) Therefore, the bulkiness of the carborane cage is important for the antagonist activity. Both carboranes work as a hydrophobic cluster to fill the cavity of the ligand-binding pocket,21,22) it is also applied to drug design. However, the molecular mechanical force field parameters of boron, which are essential for the in silico drug design for carborane, were not sufficiently developed. In the above studies, boron atoms are substituted with carbon in the docking simulation using FlexX,21) and the hydrophobicity of boron is disregarded in the calculation using Gold.20,22) Therefore, previous studies have been limited to estimating the rough complex structures only, and its accuracy is considered to be insufficient to evaluate the details, for example the calculations of binding affinity. To estimate detailed structures, the refinement of the complex structure by molecular dynamics (MD) simulation is required; however, it is impossible without the development of force field parameters. In this study, we determined the force filed parameters of p-carboranes and its substituent products to perform molecular mechanical calculations so that new carborane-based ligands can be designed in silico. The parameters were determined for each 10-vertex and 12-vertex carborane. Because we have previously determined the parameters of heme iron,23) thioester,24) and nitoryl nitroxide derivatives25) by parameter fitting using energy profiles obtained from quantum chemical calculations, those same methods were repeated in this study. Furthermore, MD simulations of AR-carborane complexes were conducted to test the determined parameters, and results were compared with structures of ligand-free AR and AR-DHT.
MATERIALS AND METHODS
Determination of Force Field Parameters for CarboraneThe force field parameters of 12-vertex and 10-vertex p-carborane (Fig. 1) were determined using the energy function of the AMBER force field,26,27) which is one of the most popular molecular mechanical force field in biological research. In the AMBER force field, the energy of the system is calculated using the energy function (Eq. 1)
 | (1) |
In this equation, r, θ, φ, and Rij are the structural features of the system (bond length, bond angle, torsional angle, and the distance between atoms i and j, respectively). The first and second terms, which are related to bond stretching and angle bending, respectively, express harmonic-oscillator approximations. The parameters req and θeq are the equilibrium bond length and bond angle, respectively, and Kr and Kθ are force constants. The third term is the torsional term, where Vn is the energy barrier of torsional motion, n is the periodicity, and γ is the phase. The last term represents the nonbonding, Coulombic, and van der Waals interactions. Here qi and qj are the atomic charges of atoms i and j, respectively. The total charge is divided into partial charges assigned to all atoms in the molecule, and the electrostatic energies are calculated based on these atomic charges using the Coulomb equation. For the calculations of van der Waals interactions, 6–12 terms were used. The residual portion of the energy that cannot be evaluated by Coulombic and van der Waals terms was corrected by the torsional term.
For parameter fitting, energy profile calculations were conducted for model molecules including carboranes with substituents R, which are -CH3, -CH2CH3, -C6H5, -CH2OH (Fig. 2A). In this figure, newly defined atom types B1–B4, and Cb are illustrated. Although all boron was regarded with the same properties, binding in the toward the equator (for example, B1, B2) and in the meridian direction (for example, B1–B4) should be distinguished for both the 12-vertex and 10-vertex carboranes. As atomic charges, restrained electrostatic potential (RESP) charges,28) which are recommended for the AMBER calculations, were used. To determine the parameters for van der Waals terms, energy profiles when methane or benzene approached the carborane were calculated using single-point energy calculations. Two approaching ways were examined (Fig. 2B). As shown in Fig. 2B, methane or benzene moved along carbon–carbon axis in the way “I” and in the way “II” methane or benzene approached the midpoint of boron–boron bond along the perpendicular of the carbon–carbon axis. In these calculations, benzene was fixed in a direction perpendicular to the carbon–carbon axis, and methane was fixed with face of the tetrahedron oriented toward the carborane cage. The energy profiles of bond stretching and angle bending for the substituents, which were located at the outer side of the polyhedral carborane structure, were determined by changing only the corresponding bond or angle or both using the same methods in Refs. 23–25. In the structural change involving the polyhedral structure, since a plurality of bond lengths, angles, and torsional angles are varied, the parameters should be calculated simultaneously using multiple regression. Three types of bond-stretching parameters must be determined for the polyhedral structure, that is, Cb-B1, B1-B2, and B1-B4. These bond-stretching parameters were obtained using the energy profiles for the expansion and contraction of carborane, in which only the bond lengths changed while maintaining all angles (Fig. 2C). After the parameter determination, other parameters related to the polyhedral structures were calculated by moving the partial structures of model molecules.
To calculate the force-field parameters, the energy profiles obtained from quantum chemical calculation were used. The structures of the carborane derivatives were optimized by B3LYP/6-311+G(2d, p), and the RESP charge was calculated using HF/6-31G(d), which is the recommended method for the RESP calculations of the AMBER force field.29) For calculating energy profiles, MP2/6-311+G(2df, p) was used. All quantum chemical calculations were performed using the Gaussian09 software.30)
Molecular Dynamics Simulation for Parameter TestThe ligand structures used in the test simulations are shown in Fig. 3. The initial structures of AR were generated based on the experimental structure registered in the Protein Data Bank (PDB). Ligand-free AR structure was constructed from the complex structure of AR and Metribolone (R1881), which is an AR agonist (PDB ID: 1E3G),31) by removing R1881 and water molecules. The initial structure of AR-DHT complex was generated from the crystal structure of AR-DHT complex (PDB ID: 2AMA).32) The disordered residues in 2AMA were modeled using the coordinates of 1E3G, and water and sulfuric acid molecules were eliminated. The structures of AR-carborane derivatives complex were constructed by computational docking trials described in Ref. 21. The carborane derivatives, BA341, which is 12-vertex p-carborane with hydroxymethyl and benzonitrile, and 10-vertex p-carborane with the same functional groups as BA341 (10cb) were used for the parameter validation. The system was solvated using the TIP3P model,33) neutralized by the addition of a chloride ion, and calculated under the periodic boundary condition. The cutoff distance was set at 10 Å, and the particle mesh Ewald method was used for calculating the electrostatic interactions.34) The MD simulations were performed in four stages of (1) energy minimization of water and ions, (2) energy minimizations of the entire system, (3) temperature-increasing MD simulation, and (4) equilibrating MD simulations. The minimizations in (1) and (2) were performed in 1000 and 2500 steps, respectively. Temperature-increasing MD simulations were performed for 20 ps with the temperature raised from 0 to 300 K at 1 bar, and then equilibrating MD simulations were performed under constant temperature and pressure. The simulation times for equilibrating MD simulations were 100 ns. The time steps of (3) and (4) were 2 fs. After those simulations, 2500 steps energy minimization was performed for all atoms to obtain final structures. Those MD simulations were accomplished using AMBER16.27) The AMBER force field (AMBER ff14SB) and general AMBER force filed (GAFF)35) were used for the parameters of amino acids and DHT, respectively.
RESULTS AND DISCUSSION
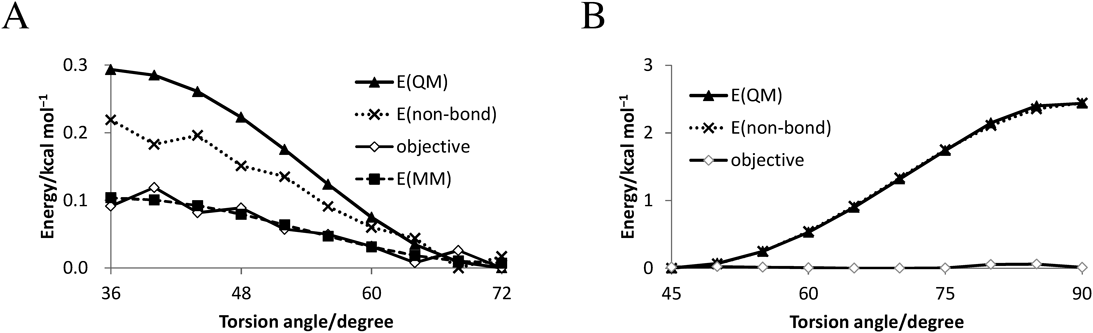
Development of Force Filed of CarboraneThe force field parameters were determined by the fittings for the energy profiles. For example, the energy profiles of B-Cb-c3-c3 and B-Cb-ca-ca torsional rotation are illustrated in Fig. 4. In this figure, energies by quantum chemical calculations E(QM), the nonbonding term E(nonbonding), differences between E(QM) and E(nonbonding) (objective), and the energies calculated by obtained parameters E(MM) are shown. E(QM)–E(nonbonding) is an objective variable for the parameter fitting of the torsional angle term (Fig. 4A). In the fitting of E(MM) for the R = –CH2CH3 derivative, the residual sum of square was 1.15 × 10−3 kcal2 mol−2. Alternatively, the objective in R = –C6H5 derivative was nearly zero because E(QM) and E(nonbonding) had approximately equal values in the structural change rotated around the Cb-ca bond (Fig. 4B). Therefore, the parameters for X-Cb-ca-X torsional rotation were zero. Parameters calculated in these manners are shown in Tables 1–4. Hydrogen atom bonded to boron in carborane (Hb) and hydrogen atom bonded to carbon in carborane (Hc) are hydrogen atoms binding to boron and carbon atoms, respectively, and atoms with all lowercase letters, such as c3 and ca correspond to the atom type in GAFF. The same van der Waals parameters were used for atoms of 10- and 12-vertex carborane (Table 1). Although the structures of 10- and 12-vertex carborane are different, those parameters of bond stretching, angle bending, and torsional rotation tended to be similar values (Tables 2–4). In addition, parameters of bond stretching and angle bending in atoms constituting the polyhedral structures were smaller than those of other atoms.
Table 1. van der Waals Parameters
| R | ε |
|---|
| B | 2.285 | 0.1786 |
| Cb | 1.908 | 0.1317 |
| Hb | 1.019 | 0.015 |
| Hc | 1.019 | 0.015 |
Hb: hydrogen atom bonded to boron in carborane. Hc: hydrogen atom bonded to carbon in carborane.
Table 2. Bond Stretching
| 10 Vertices | 12 Vertices |
|---|
| Kr | req | Kr | req |
|---|
| Cb-ca | 449.1 | 1.446 | 449.1 | 1.446 |
| Cb-c3 | 284.0 | 1.491 | 284.0 | 1.491 |
| Cb-Hc | 419.4 | 1.085 | 419.4 | 1.085 |
| B-Hb | 298.2 | 1.184 | 298.2 | 1.184 |
| B-Cb | 115.6 | 1.602 | 105.8 | 1.687 |
| B1-B2 | 117.9 | 1.869 | 97.98 | 1.742 |
| B1-B4 | 117.8 | 1.800 | 97.98 | 1.766 |
Table 3. Angle Bending
| 10 Vertices | 12 Vertices |
|---|
| Kθ | θeq | Kθ | θeq |
|---|
| ca-ca-Cb | 112.532 | 117.0 | 157.819 | 115.6 |
| ca-Cb-B | 8.253 | 124.4 | 0 | 117.9 |
| c3-Cb-B | 27.827 | 124.8 | 17.281 | 117.6 |
| Cb-c3-hc | 39.263 | 108.3 | 35.224 | 105.6 |
| Cb-c3-oh | 122.684 | 101.7 | 135.434 | 100.3 |
| B-Cb-Hc | 23.469 | 125.5 | 22.498 | 117.0 |
| B1-Cb-B2 | 83.686 | 70.1 | 16.295 | 66.4 |
| B1-B2-Cb | 83.686 | 57.1 | 16.295 | 57.3 |
| B1-B2-Hb | 17.641 | 117.3 | 17.334 | 119.7 |
| B1-B2-B4 | 12.594 | 65.1 | 12.523 | 56.5 |
| B1-B2-B3 | 0 | 90 | 32.012 | 107.5 |
| B1-B4-B2 | 12.594 | 60.2 | 12.523 | 66.5 |
| B1-B4-Hb | 17.641 | 113.6 | 17.334 | 123.2 |
| B1-B4-Cb | 0 | 107.6 | 0 | 104.8 |
| Cb-B-Hb | 25.148 | 117.2 | 20.387 | 119.4 |
| B1-Cb-B3 | 44.186 | 117.2 | 38.492 | 114.5 |
| B3-B2-B4 | 0 | 102 | 0 | 107.9 |
Table 4. Torsional Rotation
| No. | Vn /2 | γ | n |
|---|
| X-ca-Cb-X | 1 | 0 | 0 | 0 |
| X-Cb-B-X | 1 | 0 | 0 | 0 |
| B-Cb-c3-hc | 1 | 0 | 0 | 0 |
| X-B-B-X | 1 | 0 | 0 | 0 |
| B-Cb-c3-oha) | 1 | 0.08045 | 180.0 | 4.0 |
| B-Cb-c3-c3a) | 1 | 0.04907 | 180.0 | 4.0 |
| B-Cb-c3-ohb) | 1 | 0.08136 | 180.0 | 4.0 |
| B-Cb-c3-c3b) | 1 | 0.01929 | 180.0 | 4.0 |
a) 10-vertex carborane b) 12-vertex carborane.
MD simulations for ligand-free AR, AR-DHT, AR-BA341, and AR-10cb complexes were performed to evaluate the force field parameters determined in this study and to investigate the effects of carborane binding on AR structures. The ligand structures are shown in Fig. 3. BA341, which has a high AR antagonist activity,21) is a 12-vertex para-carborane with hydroxymethyl and benzonitrile as substituents. The compound 10cb, which has the same functional groups as BA341, is a partial AR agonist. For ligand-free AR and each complex, MD simulations were performed for 100 ns, and the root mean square deviations (RMSDs) are shown in Fig. 5. The RMSD values in ligand-free AR, AR-DHT, and AR-BA341 were almost constant, and these structures are considered to be converged by the 100 ns simulations; therefore, huge structural changes by ligand removal and carborane binding in these simulations did not occur. In the AR-10cb complex, although the RMSD value gradually increased by approximately 50 ns in comparison with that of other complexes, its value was considered to be converged at the end of the simulation. Figure 6 shows the AR structures at the end of the simulations, and the H1–H12 are indicated. All ligands remained in the ligand-binding pocket, and the helix structures of the H1–H12 were maintained, except for the end of H10 in the AR-DHT complex. Since helix collapse oh H10 was observed in some crystal structures with agonists or antagonists, for example 5JJM, 5VO4, and 4QL8, it is assumed to be unnecessary for agonist/antagonist activities. To investigate the influence of ligand-binding on the three-dimensional (3D) structures of AR, the root mean square fluctuations (RMSFs) of each Cα atom in the last 10 ns were calculated (Fig. 7). In ligand-free AR, the RMSF of residues of 669–700 and 842–855 included in the H1–H3 and H9–H10, respectively, loops were high, and they were more flexible than other residues of AR. RMSF values of those loops in the AR-DHT and AR-BA341 were slightly lower than those in ligand-free AR and AR-10cb. These results suggest that agonist/antagonist binding decreases AR structural flexibility regardless of the ligand function. In particular, the decrease in RMSF values of residues contained in the H9–H10 loop by antagonist binding has also been reported in another computational research.36) The effects of ligand-binding on AR flexibility are presumed to be caused by changes in the interaction between the residues in the vicinity of this loop. In Table 5, hydrogen-bond frequencies in the N-terminal side of H10 and H10–C-terminal region are shown. As described in this table, hydrogen-bond frequencies for Thr850–Arg854 and Ser851–Arg855 in the H10 were affected by bound ligands (Figs. 8A–D). The hydrogen bond between the main chain oxygen of Thr850 and main chain hydrogen of Arg854 formed over 90% of frames in the AR-DHT and AR-BA341, but the percentage of hydrogen-bond formations were 64.5 and 41.6% in the ligand-free AR and AR-10cb, respectively. In contrast, the hydrogen-bond frequencies between the main chain oxygen of Ser851 and the main chain hydrogen Arg855 were approximately 90% in the ligand-free AR, AR-DHT, and AR-BA341, and that of only 10cb was relatively low (65.1%). These differences may affect the 3D structure of H10 and interactions between the H10 and C-terminal loops. In fact, the hydrogen-bond frequency between the amide hydrogen of Gln867 and the main chain oxygen of Ile914 in ligand-free AR was low in contrast with those of AR-DHT and AR-BA341, while the hydrogen bond between Gln867 and Ile914 was not observed in most part of the simulation for AR-10cb (Fig. 8E, Table 5). These differences in hydrogen bonds for the H10 and H10–C-terminal region were considered to be involved in the activities of the ligand.
Table 5. Hydrogen-Bond Frequencies of the H10 and H10–C-Terminal Loops
| Hydrogen bond | Percent of hydrogen-bond formation |
|---|
| Ligand-free | DHT | BA341 | 10cb |
|---|
| Thr850 O–Arg854 H | 64.5 | 94.7 | 84.6 | 41.6 |
| Ser851 O–Arg855 H | 86.8 | 90.5 | 93.8 | 65.1 |
| Gln867 H–Ile914 O | 6.6 | 31.4 | 23.8 | 0.5 |
The ligand-binding for AR causes conformational change of H12, and it is important for regulating transcriptional activity; therefore, we focused on the structural differences in H12. Although the helix structures of H12 in ligand-free AR, AR-BA341, and AR-10cb were similar, the midpoint of H12 in the AR-DHT complex slightly shifted in comparison with others (Fig. 9A). The detailed structural differences are shown in Figs. 9B–E. The locations of Val901 were shifted, and the hydrogen bonds of the main chain were changed. In the AR-DHT complex, the amide hydrogen and the oxygen of Val901 formed hydrogen bonds with the oxygen of Ile898 and amide hydrogen of Lys905, respectively. Alternatively, in the ligand-free AR, AR-BA341, and AR-10cb, the hydrogen bonds for Ile898-Val901 and Val901-Lys905 were impaired. The amide hydrogen of Val901 formed a hydrogen bond with the amide oxygen of Glu897, but the amide oxygen of Ile898 formed no hydrogen bond in the final structures of the ligand-free AR, AR-BA341, and AR-10cb.
The agonist/antagonist binding changes the binding affinity of coactivators for AR; furthermore, Val716, Lys720, Arg726, Gln733, Met734, Gln738, Glu893, Met894, Glu897, and Ile898 in the AF-2 site of AR have been reported to be important in the interactions with coactivators.37,38) The location of these residues is shown in Fig. 10. In recent computational study, bicalutamide, an antagonist for AR, was suggested to induce the movement of positive charged region consisting of Lys720 and Gln733.37) In this report, the nitrogen atom of the Lys720 side chain was located outside the AF-2 in the agonist-binding state, but its location was relatively close to Gln733 in the antagonist-binding state. To investigate whether interactions between these residues were altered by ligand-binding, the hydrogen bonds between the side chains of Gln733 and Lys720 were extracted (Table 6). One hydrogen bond in each of the AR-DHT and AR-BA341 was detected, and the frequencies were 18.2 and 53.7%, respectively. Hence, in comparison with the agonist form, the antagonist binding is considered to cause the conformational change of the Lys720 side chain, and facilitate closing of Lys720 to Gln733. In ligand-free AR and AR-10cb, hydrogen bonds between the side chain of Lys720 and Gln733 or any other residues were not detected. These results suggest that the binding affinity of the coactivator with AR is decreased by locked Lys720 near the Gln733 in the antagonist-binding state or unrestricted Lys720 in the ligand-free and partial agonist-binding state. In a previous study, it has been suggested that the hydrogen bond between these residues is disrupted in the coactivator-binding structures.38) Therefore, for coactivator binding, it is important that the side chain of Lys720 is located in the vicinity of Gln733 and their interaction is not too strong. Alternatively, the alteration in the negative charged region of the coactivator-binding site, which is composed of the side chains of Glu893 and Glu897, could not be elucidated in the present simulations. Because all hydrogen bonds involved in their side chains are infrequently detected, they are considered to be highly flexible. To reveal the effects of carborane on the conformational changes of these residues, an extension of the simulation time or experimental validation is required.
Table 6. Hydrogen-Bond Frequencies between Gln733 and Lys720
| Hydrogen bond | Percent of hydrogen-bond formation |
|---|
| Ligand-free | DHT | BA341 | 10cb |
|---|
| Gln733 O–Lys720 H | 0 | 18.2 | 53.7 | 0 |
CONCLUSION
In this study, we developed the force field parameter of the boron cluster–carborane, which is a novel hydrophobic core using quantum chemical calculations. The coefficient of bond stretching and angle bending in the polyhedral structure tends to be small. This tendency is considered to be attributed to the dispersion of each energy term by numerous bonds and bond angles in these polyhedral structures. The parameter values are similar between 10- and 12-vertex carborane; therefore, the parameters determined in this study are assumed to be valid. These determined parameters enable the in silico drug design of carborane derivatives using the classical mechanical approximation methods. In addition to the development of the force field parameter, we performed MD simulation of AR-carborane complexes using these parameters to examine the parameters and investigate the effects of carborane binding on the AR structure by comparison with ligand-free AR and AR-DHT complexes. Those calculated results suggested that BA341 binding attenuates some hydrogen bonds involved in the structure and location of H12, which is important for the conversion between AR agonist and antagonist form.2,5,6) Because the differences in helix structure of H12 were observed between the AR-DHT complex and others, the binding of carborane derivatives are considered to impair agonist formation. Furthermore, the difference in the hydrogen bond between Lys720 and Gln733, which are important residues for interaction with the coactivator, was observed. In particular, an increase in the hydrogen-bond frequency by BA341 binding was expected to result in an alteration of the direction of the Lys720 side chain. This conformational change in Lys720 is considered to impair the coactivator binding of AR. Therefore, our computational results consisted of the experimental and computational results of previous studies and explained the mechanisms of BA341 antagonist activity. In addition to this, the force field parameter determined in this study is considered to be appropriate for structure-based drug design. Alternatively, the mechanism of partial agonist activity for 10cb is unclear. Although we have reported that partial agonist-binding estrogen receptors are often observed as an antagonist form,39) structural change in AR-10cb was not the same as that in AR-BA341. In the AR-10cb complex, the RMSD and RMSF values were relatively large in comparison with those of other complexes. These results indicate that further simulations may be needed to investigate the large structural change such as the rearrangement of H12 although a local metastable structure was obtained in this study. At least, a significant extension of the simulation time is required for accurate analysis of the structural change of AR by partial agonist binding. A longer simulation is also required for more detailed investigations of the structural influence of carborane derivatives on AR. In addition, there are some difficult points to accurately analyze the interaction between the carborane compounds and AR using a force field, such as dispersion force and charge transfer. Therefore, such points are needed to be included in the future analysis.
In summary, the parameters of p-carborane for molecular mechanical calculation such as classical MD simulations were determined, and a test of MD simulations using AR complexes was performed. This parameter is available for refining carborane-receptor structure obtained by docking simulations, and it is expected that the development of new ligands with carborane cages for various receptors are accelerated.
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research (17K08257) from the Japan Society for the Promotion of Science.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Bregadze VI. Dicarba-closo-dodecaboranes C2B10H12 and their derivatives. Chem. Rev., 92, 209–223 (1992).
- 2) Endo Y, Iijima T, Yamakoshi Y, Yamaguchi M, Fukasawa H, Shudo K. Potent estrogenic agonists bearing dicarba-closo-dodecaborane as a hydrophobic pharmacophore. J. Med. Chem., 42, 1501–1504 (1999).
- 3) Ogawa T, Ohta K, Yoshimi T, Yamazaki H, Suzuki T, Ohta S, Endo Y. m-Carborane bisphenol structure as a pharmacophore for selective estrogen receptor modulators. Bioorg. Med. Chem. Lett., 16, 3943–3946 (2006).
- 4) Kaise A, Ohta K, Endo Y. Novel p-carborane-containing multitarget anticancer agents inspired by the metabolism of 17β-estradiol. Bioorg. Med. Chem., 25, 6371–6378 (2017).
- 5) Calabrese G, Daou A, Barbu E, Tsibouklis J. Towards carborane-functionalised structures for the treatment of brain cancer. Drug Discov. Today, 23, 63–75 (2018).
- 6) Yamaotsu N, Oda A, Hirono S. Determination of ligand-binding sites on proteins using long-range hydrophobic potential. Biol. Pharm. Bull., 31, 1552–1558 (2008).
- 7) Oda A, Yamaotsu N, Hirono S. Evaluation of the searching abilities of HBOP and HBSITE for binding pocket detection. J. Comput. Chem., 30, 2728–2737 (2009).
- 8) Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, Blumberg B, Kastner P, Mark M, Chambon P, Evans RM. The nuclear receptor superfamily: the second decade. Cell, 83, 835–839 (1995).
- 9) Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr. Rev., 23, 175–200 (2002).
- 10) Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr. Rev., 25, 276–308 (2004).
- 11) Gobinet J, Poujol N, Sultan Ch. Molecular action of androgens. Mol. Cell. Endocrinol., 198, 15–24 (2002).
- 12) Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Curr. Opin. Cell Biol., 10, 384–391 (1998).
- 13) Tan MH, Li J, Xu HE, Melcher K, Yong EL. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin., 36, 3–23 (2015).
- 14) Dehm SM, Tindall DJ. Molecular regulation of androgen action in prostate cancer. J. Cell. Biochem., 99, 333–344 (2006).
- 15) Mohammad OS, Nyquist MD, Schweizer MT, Balk SP, Corey E, Plymate S, Nelson PS, Mostaghel EA. Supraphysiologic testosterone therapy in the treatment of prostate cancer: models, mechanisms and questions. Cancers, 9, 166 (2017).
- 16) Jernberg E, Bergh A, Wikström P. Clinical relevance of androgen receptor alterations in prostate cancer. Endocr. Connect., 6, R146–R161 (2017).
- 17) Fujimura T, Takayama K, Takahashi S, Inoue S. Estrogen and androgen blockade for advanced prostate cancer in the era of precision medicine. Cancers, 10, 29 (2018).
- 18) Speers C, Zhao SG, Chandler B, Liu M, Wilder-Romans K, Olsen E, Nyati S, Ritter C, Alluri PG, Kothari V, Hayes DF, Lawrence TS, Spratt DE, Wahl DR, Pierce LJ, Feng FY. Androgen receptor as a mediator and biomarker of radioresistance in triple-negative breast cancer. NPJ Breast Cancer, 3, 29 (2017).
- 19) Fujii S, Goto T, Ohta K, Hashimoto Y, Suzuki T, Ohta S, Endo Y. Potent androgen antagonists based on carborane as a hydrophobic core structure. J. Med. Chem., 48, 4654–4662 (2005).
- 20) Ohta K, Ogawa T, Kaise A, Oda A, Endo Y. Aliphatic substitution of o-carboranyl phenols enhances estrogen receptor beta selectivity. Chem. Pharm. Bull., 62, 386–391 (2014).
- 21) Ohta K, Goto T, Fujii S, Kawahata M, Oda A, Ohta S, Yamaguchi K, Hirono S, Endo Y. Crystal structure, docking study and structure–activity relationship of carborane-containing androgen receptor antagonist 3-(12-hydroxymethyl-1,12-dicarba-closo-dodecaboran-1-yl) benzonitrile. Bioorg. Med. Chem., 19, 3540–3548 (2011).
- 22) Fujii S, Ohta K, Goto T, Oda A, Masuno H, Endo Y, Kagechika H. Development of androgen receptor ligands by application of ten-vertex p-carborane as a novel hydrophobic core structure. Med. Chem. Commun., 3, 680–684 (2012).
- 23) Oda A, Yamaotsu N, Hirono S. New AMBER force field parameters of heme iron for cytochrome P450s determined by quantum chemical calculations of simplified models. J. Comput. Chem., 26, 818–826 (2005).
- 24) Oda A, Fukuyoshi S, Nakagaki R, Takahashi O. Determination of AMBER force field parameters for thioester by quantum chemical calculations. Chem. Lett., 42, 1206–1208 (2013).
- 25) Oda A, Fukuyoshi S, Kurimoto E. Determination of molecular force field parameters for nitronyl nitroxide derivatives using quantum chemical calculations. Polyhedron, 136, 93–101 (2017).
- 26) Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput., 11, 3696–3713 (2015).
- 27) Case DA, Babin V, Berryman JT, et al. Amber 16, University of California, San Francisco (2014).
- 28) Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J. Phys. Chem., 97, 10269–10280 (1993).
- 29) Wang J, Cieplak P, Kollman PA. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem., 21, 1049–1074 (2000).
- 30) Frisch MJ, Trucks GW, Schlegel HB, et al. Gaussian 09, Revision C.01, Gaussian Inc., Wallingford (2010)
- 31) Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, Bäsler S, Schäfer M, Egner U, Carrondo MA. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J. Biol. Chem., 275, 26164–26171 (2000).
- 32) Pereira de Jésus-Tran K, Côté PL, Cantin L, Blanchet J, Labrie F, Breton R. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci., 15, 987–999 (2006).
- 33) Joung IS, Cheatham TE III. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B, 112, 9020–9041 (2008).
- 34) Darden T, York D, Pedersen L. Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J. Chem. Phys., 98, 10089–10092 (1993).
- 35) Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J. Comput. Chem., 25, 1157–1174 (2004).
- 36) Sakkiah S, Kusko R, Pan B, Guo W, Ge W, Tong W, Hong H. Structural changes due to antagonist binding in ligand binding pocket of androgen receptor elucidated through Molecular Dynamics simulations. Front. Pharmacol., 9, 492 (2018).
- 37) Estébanez-Perpiñá E, Arnold LA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, Webb P, Fletterick RJ. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc. Natl. Acad. Sci. U.S.A., 104, 16074–16079 (2007).
- 38) Hsu CL, Liu JS, Wu PL, Guan HH, Chen YL, Lin AC, Ting HJ, Pang ST, Yeh SD, Ma WL, Chen CJ, Wu WG, Chang C. Identification of a new androgen receptor (AR) co-regulator BUD31 and related peptides to suppress wild-type and mutated AR-mediated prostate cancer growth via peptide screening and X-ray structure analysis. Mol. Oncol., 8, 1575–1587 (2014).
- 39) Kato K, Fujii K, Nakayoshi T, Watanabe Y, Fukuyoshi S, Ohta K, Endo Y, Yamaotsu N, Hirono S, Kurimoto E, Oda A. Structural differences between the ligand-binding pockets of estrogen receptors alpha and beta. J. Phys. Conf. Ser., 1136, 012021 (2018).