Notes
KB-R7943 Inhibits the Mitochondrial Ca2+ Uniporter but Not Na+–Ca2+ Exchanger in Cardiomyocyte-Derived H9c2 Cells
2020 Volume 43 Issue 12 Pages 1993-1996

Details
2020 Volume 43 Issue 12 Pages 1993-1996
The effect of KB-R7943, an inhibitor of the plasmalemmal Na+–Ca2+ exchanger, on mitochondrial Ca2+ transporters was examined with membrane-permeabilized cardiomyocyte-derived H9c2 cells expressing the fluorescent Ca2+ indicator, yellow cameleon 3.1, in the mitochondria. KB-R7943, as well as ruthenium red, inhibited the rise in mitochondrial Ca2+ on increasing the extramitochondrial Ca2+ concentration from 0 nM to 300 nM. CGP-37157, but not KB-R7943, inhibited the decline in mitochondrial Ca2+on return to Ca2+ free extramitochondrial solution. These results indicated that KB-R7943 has inhibitory effects on the mitochondrial Ca2+ uniporter, but not on the mitochondrial Na+–Ca2+ exchanger.
The Na+–Ca2+ exchanger (NCX) is involved in the regulation of Ca2+ concentration in the myocardium. The plasmalemmal NCX basically functions in the forward (Ca2+ efflux) mode, but under pathological conditions such as ischemia-reperfusion, it functions in the reverse (Ca2+ influx) mode and causes cellular damage through cellular Ca2+ overload.1) The two major NCX inhibitors with benzyloxyphenyl structure, KB-R7943 (2-[2-[4-(4-nitrobenzyloxy)phenyl]ethyl]isothiourea methanesulfonate) and SEA0400 (2-[4-[(2,5-difluorophenyl)methoxy]phenoxy]-5-ethoxyaniline), have been used as potent pharmacological tools in NCX research.2) Earlier studies have shown that NCX inhibitors have protective effects against ischemia–reperfusion injury in several organs including the heart. In models of myocardial ischemia–reperfusion, KB-R7943 and SEA0400 reduced cytoplasmic and mitochondrial Ca2+ overload and enhanced the recovery of contractile force and ATP content3–6); this could be explained by reduction of Ca2+ entry into the cytoplasm through plasmalemmal NCX. These results suggested that inhibition of plasmalemmal NCX protects the mitochondria through attenuation of cytoplasmic Ca2+ overload during myocardial ischemia.
Another possible explanation for the mitochondrial protective effects of NCX inhibitors was their direct action on mitochondrial Ca2+ transport, either reduction of Ca2+ entry or enhancement of Ca2+ extrusion. To examine such possibility, we established cardiomyocyte-derived H9c2 cells expressing the Ca2+ sensitive fluoroprobe yellow cameleon 3.1 in the mitochondria, which enabled observation of mitochondrial Ca2+ entry and Ca2+ extrusion. Using this system, we previously reported that SEA0400 affects neither mitochondrial Ca2+ entry nor Ca2+ extrusion.7,8) Concerning the effect of KB-R7943 on mitochondrial Ca2+ transporters, conflicting results have been reported depending on the cell type.9,10) In the present study, we intended to clarify the effect of KB-R7943 on mitochondrial Ca2+ transport in cardiomyocytes using the H9c2 cell system described above.
The experimental system was basically the same as that described in our previous reports.7,8) Rat embryonic heart derived H9c2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). To obtain mitochondrial-targeted yellow cameleon DNA, its full-length cDNA11) was inserted into an expression vector which has a mitochondria targeting signal sequence, pEYFP-mito (Clontech, Palo Alto, CA, U.S.A.). This vector was introduced into H9c2 cells with lipofectamine 2000 (Invitrogen, Carlsbad, CA, U.S.A.) and stable transformants were obtained by clone culture in the presence of G418, a neomycin analogue. For the measurement of mitochondrial function, the plasmalemmal membrane was permeabilized by perfusion of digitonin (20 µg/mL) in a Ca2+-free solution that contained (in mM) 50 KCl, 80 potassium aspartate, 4 sodium pyruvate, 20 N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), 3 MgCl2, 3 Na2ATP, 5.8 glucose, and 3 ethylene glycol bis(2-aminoethyl ether)-N,N,N′,N′-tetraacetic acid (EGTA) (pH 7.3 with KOH). The free Ca2+ concentration in the extramitochondrial solution was changed to 300 nM according to the experimental protocol. The cells were observed with an epifluorescence microscope (IX70, Olympus, Tokyo, Japan). The cells were excited at 440 nm and the emissions at 480 ± 30 and 535 ± 25 nm were detected by a cooled CCD camera (HISCA, Hamamatsu Photonics, Shizuoka, Japan) at a time resolution of 5 s, and ratioed after correction of background fluorescence (Aquacosmos software, Hamamatsu Photonics). The inhibitors used were KB-R7943 (Nihon Organon, Osaka, Japan), ruthenium red (Sigma, St. Louis, MO, U.S.A.), and CGP-37157 (Tocris, Bristol, U.K.). All data were expressed as means ± standard error of the mean (S.E.M.). Data were analyzed by the one-way ANOVA followed by Dunnett’s multiple tests. If the p value was less than 0.05, the difference was considered statistically significant.
In permeabilized H9c2 cells, raising the extramitochondrial Ca2+ concentration from 0 nM to 300 nM resulted in an increase in fluorescence ratio, which indicated an increase in mitochondrial Ca2+ concentration. On return to Ca2+ free extramitochondrial solution, the fluorescence ratio declined toward its initial value indicating a decrease in mitochondrial Ca2+ concentration. The rise and fall of fluorescence ratio was reproducible which enabled evaluation of pharmacological effects.
The increase in fluorescence ratio caused by raising the extramitochondrial Ca2+ concentration from 0 nM to 300 nM was used as an index of mitochondrial Ca2+ uniporter activity (Fig. 1). Ruthenium red (1 µM), an inhibitor of the mitochondrial Ca2+ uniporter, had no effect on the fluorescence ratio under Ca2+ free extramitochondrial solution. The rise in fluorescence ratio by 300 nM extramitochondrial Ca2+ was markedly inhibited by ruthenium red, which confirmed the involvement of the Ca2+ uniporter. KB-R7943 (10 µM) had no effect on the fluorescence ratio under Ca2+ free extramitochondrial solution, but significantly inhibited the rise in fluorescence ratio by 300 nM extramitochondrial Ca2+.
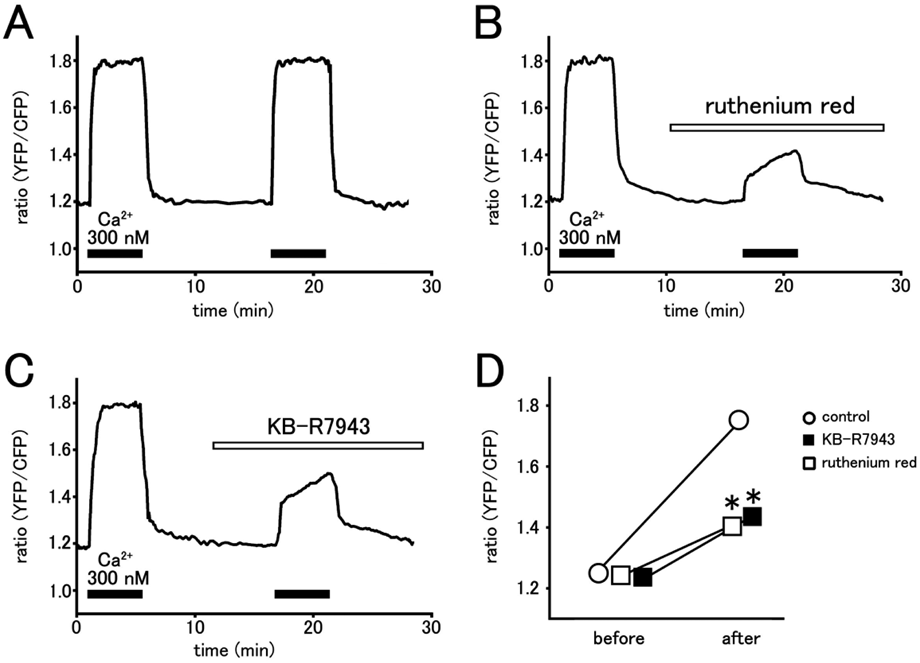
The H9c2 cells expressing mitochondria-targeted yellow cameleon 3.1 were permeabilized and fluorescence ratio was measured as an index of intramitochondrial Ca2+ concentration. The cells were kept in a Ca2+ free extramitochondrial solution and exposed to extramitochondrial solution containing 300 nM Ca2+ for 5 min twice with a 10 min interval. Before the second elevation of Ca2+, the cells were untreated with agents (control; A), treated with 1 µM ruthenium red (B) or treated with 10 µM KB-R7943 (C). Summarized results of fluorescence ratio before and after the second application of 300 nM Ca2+ were presented (D). Data points with vertical bars indicate the mean ± standard error of the mean (S.E.M.) from 4–6 cells. Asterisks indicate significant differences (p < 0.05) from the corresponding control values.
The decline in fluorescence ratio on return to Ca2+ free extramitochondrial solution was used as an index of the mitochondrial NCX activity (Fig. 2). CGP-37157 (10 µM), an inhibitor of the mitochondrial NCX, had no effect on the fluorescence ratio under 300 nM extramitochondrial Ca2+. The decline in fluorescence ratio on return to Ca2+ free extramitochondrial solution was markedly inhibited by CGP-37157, which confirmed the involvement of mitochondrial NCX. KB-R7943 (10 µM) affected neither the fluorescence ratio under 300 nM extramitochondrial Ca2+ nor the decline in fluorescence ratio on return to Ca2+ free extramitochondrial solution.

The permeabilized H9c2 cells were kept in a Ca2+ free extramitochondrial solution and exposed to extramitochondrial solution containing 300 nM Ca2+ for 5 min. Before return to 0 nM Ca2+ extramitochondrial solution, the cells were untreated with agents (control; A), treated with 10 µM CGP-37157 (B) or treated with 10 µM KB-R7943 (C). Summarized results of fluorescence ratio before and after the application of Ca2+ free extramitochondrial solution were presented (D). Data points with vertical bars indicate the mean ± S.E.M. from 5–6 cells. Asterisks indicate significant differences (p < 0.05) from the corresponding control values.
The present study was undertaken to clarify whether KB-R7943 has direct effects on mitochondrial Ca2+ transport in cardiomyocytes. We used membrane-permeabilized cardiomyocyte-derived H9c2 cells expressing the fluorescent Ca2+ indicator, yellow cameleon 3.1 in the mitochondria, which was proven to be useful for the evaluation of the mitochondrial effects of SEA0400.7,8)
The mitochondrial Ca2+ uniporter is considered to be the main system for Ca2+ influx into the mitochondria. This transporter is inhibited by ruthenium red or its derivative Ru360.12) Our previous study with H9c2 cells indicated that the elevation of mitochondrial Ca2+ concentration caused by an increase in extramitochondrial Ca2+ concentration was inhibited by ruthenium red and carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) which eliminates the driving force of the Ca2+ uniporter, but enhanced by CGP-37157, a mitochondrial NCX inhibitor.8) In our preliminary experiments, cyclosporine A, which inhibits the opening of the mitochondrial permeability transition pore (PTP), had no effect on the increase in mitochondrial Ca2+. These results indicated that Ca2+ influx mainly occurs through the Ca2+ uniporter.
Whether KB-R7943 affects mitochondrial Ca2+ uptake is controversial; KB-R7943 inhibited the Ca2+ uniporter in Hela cells,9) while it did not in AD293 cells10); this suggests that the pharmacological property of the Ca2+ uniporter may differ among cell types. Our present results indicated that KB-R7943 inhibits the Ca2+ uniporter in H9c2 cells. We also confirmed that KB-R7943 did not depolarize the mitochondrial membrane in cardiomyocytes (data not shown). Thus, the presently observed inhibitory effect of KB-R7943 on mitochondrial Ca2+ uptake was unlikely to be an indirect effect through mitochondrial depolarization. The inhibition of mitochondrial Ca2+ uptake by KB-R7943 probably is an advantage for the treatment of ischemia–reperfusion injury. In fact, ruthenium red was reported to reduce mitochondrial Ca2+ overload and prevent cell death in cardiomyocytes.13) As KB-R7943 is membrane permeable, it is accessible to the mitochondrial Ca2+ uniporter when applied extracellularly in intact cells or tissue preparations. Thus, blockade of the mitochondrial Ca2+ uniporter probably contributes to the mitochondria protective action of KB-R7943.
The mitochondrial NCX is considered to be the main system for extrusion of mitochondrial Ca2+ into the cytoplasm. This transporter is encoded by the NCLX gene which has 62% homology with the plasmalemmal NCX in their transmembrane repeats α1 and α2.14) Mitochondrial NCX can be discriminated from plasmalemmal NCX by their pharmacological properties; while SEA0400 selectively inhibits plasmalemmal NCX, CGP-37157, a benzothiazepine compound, selectively inhibits mitochondrial NCX.7,15) The present results showed that KB-R7943 does not inhibit mitochondrial NCX. Whether this is an advantage for the treatment of ischemia–reperfusion injury awaits further investigation. It was reported in the rat heart that inhibition of the mitochondrial NCX by clonazepam was deleterious under ischemia–reperfusion.16)
The two major NCX inhibitors, KB-R7943 and SEA0400, block the forward and reverse modes of the plasmalemmal NCX, but not mitochondrial NCX. These inhibitors, especially KB-R7943, has additional blocking effect on plasmalemmal voltage dependent ion channels17) and intracellular transporters.10) In addition, the present study indicated that KB-R7943 inhibits the mitochondrial Ca2+ uniporter in cardiomyocyte-derived H9c2 cells. Such profiles should be taken into consideration when applying these NCX inhibitors to assay systems and pathological models.
The authors express their thanks to Dr. Atsushi Miyawaki (Riken Brain Science Institute, Japan) for kindly providing yellow cameleon 3.1. This work was supported by JSPS KAKENHI Grant Numbers JP20K07299, JP20K16013, and JP20K07091.
The authors declare no conflict of interest.