Abstract
The purpose of this study was to investigate the preventive effect and mechanism of Dendrobium alkaloids (DNLA) on oxidative stress-related death in neuronal cells. Our results demonstrated that DNLA has a direct neuroprotective effect through oxidative stress in N2A cells induced by hydrogen peroxide (H2O2). CCK8, lactate dehydrogenase (LDH), intracellular Ca2+, intracellular reactive oxygen species (ROS), and mitochondrial membrane potential (MMP) were used to evaluate the mechanism of DNLA neutralization by H2O2-induced injury. Results presented in the paper indicate that treatment with DNLA (35 ng/mL) significantly attenuated decreases in cell viability, release of LDH, and apoptosis after H2O2-induced neuronal injury. Furthermore, DNLA significantly reduced intracellular Ca2+ up-regulation, ROS production, and inhibited mitochondrial depolarization. Moreover, DNLA treatment significantly downregulated expressions of interleukin (IL)-1β, tumor necrosis factor (TNF)-α, IL-6, nitric oxide synthase, janus kinase–signal transducer and activators of transcription (JAK–STATs) signaling in N2A cells, all of which were H2O2-induced. Taken together, our findings suggested that DNLA may inhibit the expression of pro-inflammatory and pro-apoptotic factors by blocking JAK–STATs signaling after oxidative stress injury. This research provides a potential experimental basis for further application of DNLA to prevent various human nervous system diseases caused by oxidative stress.
INTRODUCTION
Oxidative stress is accompanied by the occurrence and development of many diseases, which can not only cause cell injury but also cause irreversible cell necrosis.1) The occurrence of oxidative stress generally means an imbalance between the oxidant and the antioxidant, leading to an excessive production of free radicals and a decrease in the dynamic detoxification ability of the reaction intermediate.2) High oxygen demand and relatively few antioxidants are the main features of the mammalian brain which are generally particularly vulnerable to oxidative damage.3,4) Free radical accumulation promotes oxidative stress by inducing cell swelling and apoptosis affects the composition of nerve cells, causing a series of neurological diseases such as Alzheimer’s disease (AD), and cerebral ischemia-reperfusion injury.5,6) For those reason, antioxidant stress could be a potential target for treating neuronal damage in stroke and other neurological diseases.7) In addition, oxidative stress, after causing brain tissue damage, could result in the expression of pro-inflammatory or pro-apoptotic factors in local tissues and cells, thereby further increasing the incidence of inflammation and apoptosis.8) These inflammatory reactions and apoptosis not only cause secondary tissue and cell damage, but also have adverse effects on the prognosis of the disease.9) Therefore, for treatment of neurological diseases caused by a series of inflammatory and apoptotic reactions and the generation of superoxide free radicals, it is the main strategy to search for neuroprotective agents.
Oxidative stress has a certain degree with inflammatory reactions and apoptosis, and those may be induced by death receptors, and endoplasmic reticulum stress.10–12) Inflammatory reactions and cell apoptosis are a hallmark of cell necrosis and severe oxidative stress.13) The results indicate that low doses of hydrogen peroxide (H2O2) can cause cell membrane lipid peroxidation and decreased cell membrane permeability and organelle damage early in the stimulation, which may lead to volume-regulated ion channel and cell membrane damage followed by inflammatory reactions and cell apoptosis.14,15) In a state of sustained oxidative stress, the damaged cells undergo inflammatory reactions, apoptosis or necrosis.16) Neuronal injury, inflammatory response, or apoptosis may exacerbate oxidative stress, further promoting cell and tissue necrosis.17) Therefore, intervention of inflammatory reactions and cell apoptosis can alleviate oxidative stress-induced neuronal damage.18)
Together, janus kinase (JAK) and signal transducer and activators of transcription (STATs), comprise the JAK–STATs pathway, which is an important inflammatory response signaling pathway that mediates immune response and cell apoptosis via H2O2-induced inducible nitric oxide synthase (iNOS) and Bax expression.19,20) In multiple cell types, H2O2 is the main factor in JAK–STATs signaling pathways activation, which acts as an intracellular signaling molecule for phosphorylation of JAK.21) JAK–STATs are the classic cascade of cell damage and a large number of studies have shown that activation of JAK–STATs regulates oxidative stress-induced neuronal damage. Phosphorylated STATs form homology or isomers heterodimer, that regulate transcription of target genes encoding pro-inflammatory cytokines, pro-apoptotic factor and inducible enzymes, including iNOS, cyclooxygenase (COX)-2, interleukin (IL)-6, Bax.22–24) There is an increasing curious about regulating JAK–STATs activity because of its role in mediating inflammation and apoptosis related gene expression.
Dendrobium nobile Lindl. alkaloid (DNLA) is a bioactive component extracted from the traditional Chinese medicine which called Dendrobium nobile Lindl.25) Previous research has demonstrated that DNLA has a variety of biological activities, such as anti-oxidation,26) anti-inflammatory,27) hepatoprotective27,28) and anti-neurodegenerative activities.29) Induction of nuclear factor-E2-related factor 2 (Nrf2) pathway by DNLA have been proven by Li et al.30) Meanwhile, DNLA is known to protect oxygen-glucose deprivation and reperfusion induced neuronal cells by increasing cell viability and decreasing cell apoptosis in vitro.31) However, the neuroprotective mechanism of DNLA on neurotoxicity is unclear. This study provided molecular-level insights into the mechanisms by which DNLA exerts anti-inflammatory and anti-apoptotic effects.
MATERIALS AND METHODS
Antibodies and ReagentsPolyclonal antibodies against JAK1, phospho-JAK1 (Tyr1034), JAK2, phospho-JAK2 (Tyr1007), STAT1, phospho-STAT1 (Tyr701), STAT3, and phospho-STAT3 (Tyr705) were provided from Cell Signaling Technology (Beverly, MA, U.S.A.). Secondary antibodies used for Western blotting were purchased from proteintech. H2O2, and dimethyl sulfoxide (DMSO) were purchased from Sigma. The CCK8 Assay Kit, lactate dehydrogenase (LDH) Cytotoxicity Assay Kit, and Fluorescence dye Fluo-4 AM were obtained from Beyotime (Shanghai, China).
Isolation, Purification and Analysis of DNLADetailed operations were performed as previously described.32)
Treatment of H2O2 and DNLANeurotoxicity was evaluated as previously described.33,34) To mimic toxic damage in vitro, cells were placed in H2O2 for 24 h at different concentrations (10, 25, 50, 100, 200 and 400 µM). DNLA was dissolved in DMSO to prepare a stock solution that was stored at −20°C. During experiments, the stock solution was diluted into three different working concentrations (1, 35, 70, 140, 280, 350, and 420 ng/mL). Cultures treated with DMSO only were used as controls.
Cell Culture and TreatmentMouse N2A neuroblastoma cells were purchased from American Type Culture Collection (ATC C, Manassas, VA, U.S.A.). Cells were supplemented in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) at full-back, streptomycin 100 µg/mL and 100 U/mL Penicillin (UT Hyclone, Logan, U.S.A.) 37°C humidifies the atmosphere with 5% carbon dioxide. To mimic the ischemic state in vitro, N2A cells were exposed to H2O2 for 24 h.
Cell Viability AssayCell viability was detected using Cell Counting Kit-8 (Beyotime). Sepecifically, cells were seeded on 96-well plates and and the cells were subsequently exposed to H2O2 treatment. Another group of cells treated with DNLA when exposure to H2O2. Following the treatment, 10 µL of CCK-8 was processed to 37°C for 2 h each incubation, and the absorbance was measured using an enzyme-linked immunosorbent assay (ELISA) reader at 450 nm. Cell viability is expressed as a percentage of the control group.
LDH Release AssayLDH released from cells 24 h after treatment was measured using an LDH Detection Kit (Beyotime). LDH release results were expressed as a percentage of the control.
Nitrite AssayNO production was calculated from the nitrite in the culture supernatant. The culture supernatant was mixed with the corresponding reagent (Beyotime), and the absorbance at 570 nm was measured using a spectrophotometer.
Determination of Intracellular Ca2+ of CellsThe intracellular Ca2+ level was measured by using the fluorescent dye Fluo-4am (Beyotime). Wash cells three times with phosphate buffered saline (PBS) filled with 10 mmol/l Fluo-4 4′-6-diamidino-2-phenylindole (DAPI) and 5 µg/mL in an incubator and incubate for 30 min in the dark at 37°C. Then, the fluorescence of Ca2+ in the cells was observed under a fluorescence microscope. Analysis of Ca2+ fluorescence intensity using Image J software (National Institutes of Health, Bethesda, MD, U.S.A.).
Determination of Mitochondrial Membrane Potential (MMP) of N2A CellsMMP was measured using JC-1 dye (Beyotime). Cells were washed the three times with PBS and incubated with 2 µm/L JC-1, 37 for 20 min. Next, JC-1 fluorescence was detected with confocal laser scanning microscopy. Under confocal laser scanning microscopy, green fluorescence showed lower cell Ψm, indicating that JC-1 maintained a monomeric form, while red fluorescence showed high cell Ψm. The relative ratio of red and green fluorescence is used to measure the degree of mitochondrial depolarization.
RNA Extraction and Quantitative (q)RT-PCRExtraction of total RNA is obtained by using TriPure Reagent (Roche Diagnostics, Indianapolis, IN, U.S.A.). Each RNA sample (10 ng) was reverse-transcribed into cDNA. RT-PCR was performed using a 7500 real-time PCR system, with AceQ qPCR SYBR Green Master Mix. RT-PCR primers were as follows: TNF-α, 5′-CCC TCA CAC TCA GAT CAT CTT CT-3′ and 5′-GCT ACG ACG TGG GCT ACA G-3′; IL-6, 5′-TAG TCC TTC CTA CCC CAA TTT CC-3′ and 5′-TTG GTC CTT AGC CAC TCC TTC-3′; IL-1β, 5′-GCA ACT GTT CCT GAA CTC AAC T-3′ and 5′-ATC TTT TGG GGT CCG TCA ACT-3′; iNOS, 5′-GTT CTC AGC CCA ACA ATA CAA GA-3′ and 5′-GTG GAC GGG TCG ATG TCA C-3′; Bax, 5′-TGA AGA CAG GGG CCT TTT TG-3′ and 5′-AAT TCG CCG GAG ACA CTC G-3′; Bcl-2, 5′-ATG CCT TTG TGG AAC TAT ATG GC-3′ and 5′-GGT ATG CAC CCA GAG TGA TGC-3′; GAPDH, 5′-AGG TCG GTG TGA ACG GAT TTG-3′ and 5′-TGT AGA CCA TGT AGT TGA GGT CA-3′. The data for each sample were normalized to GAPDH expression levels.
Western BlotWestern blot was performed as previously described.35) Briefly, prior to total protein extraction, the protein concentration of each group of samples was quantified using the BCA kit. After the samples were boiled, the samples were tested by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). The electrophoresed protein was transferred to a polyvinylidene difluoride (PVDF) membrane using a transfer device (Roche Diagnostics, Mannheim, Germany) and set at 300 mA for 90 min. The membrane is then incubated witβh the corresponding primary antibody including mouse anti-JAK1, anti-phosphorylated (p)-JAK1, anti-JAK2, anti-p-JAK2, anti-STAT1, anti-p-STAT1, anti-STAT3, anti-p-STAT3 monoclonal antibodies (dilution, 1 : 500), and mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) monoclonal antibodies, overnight at 4°C. The band after the primary antibody incubation was then incubated with horseradish peroxidase-conjugated anti-mouse immunoglobulin G (IgG) (diluted 1 : 20000) secondary antibody at room temperature for 2 h. The enhanced chemiluminescence (ECL) detection system was used for exposure, and the density signal was calculated using Image J software. The intensity of immune response bands was standardized with GAPDH corresponding bands.
Ethical ApprovalThis article does not include any research performed by any of the authors on human participants. The experiments in this article followed all applicable international, national, and institutional animal care and use guidelines.
Statistical AnalysisThe data were analyzed using SPSS version 19.0 statistical software and expressed as mean ± standard deviation. Compare differences between groups were using the ANOVA followed by Tukey’s multiple analysis. Statistically significant was set at p < 0.05.
RESULTS
DNLA Protects N2A Cells from H2O2-Induced Neuronal DamageCCK8 results confirmed that H2O2 reduced cell viability in a concentration-dependent manner (Fig. 1A). Therefore, 100 µM hydrogen peroxide treatment for 24 h is used as a condition for inducing cell damage in subsequent experiments. Treatment of N2A cells with different concentrations of DNLA (1–350 ng/mL) for 24 h did not change their viability. While, higher concentration (420 ng/mL) were found to be moderately toxic (Fig. 1B). Use of DNLA can increase H2O2-induced cell viability decline, as shown in Fig. 1C. To further study the protective effects of DNLA, the release of LDH was also measured (Fig. 1D). When treated with DNLA in N2A cells, the release of LDH was significantly reduced. Meanwhile, treatment with 35 ng/mL DNLA resulted in the most significantly inhibited H2O2-induced cell apoptosis (Fig. 1E) and NO production (Fig. 1F). These results showed that DNLA has a protective effect on H2O2-induced cytotoxicity. Thus, 35 ng/mL DNLA was used for subsequent experiments.
DNLA Reduce the Upregulation of Intracellular Ca2+ Concentration after H2O2-Induced Cell InjuryReportedly, dysregulation of intracellular Ca2+ homeostasis, one of the important mechanism undering neuronal apoptosis, participate in neuronal injury.36) To explore the role of DNLA on intracellular Ca2+ during H2O2-induced cell injury, Ca2+ concentration was measured via Fluo-4 AM dye. Compared with control or DNLA treatment group, the intracellular Ca2+ in H2O2-treated N2A cells were significantly increased. Treatment with DNLA effectively attenuated H2O2-induced upregulation of intracellular Ca2+ (Figs. 2A, B).
DNLA Relieved Mitochondrial Depolarization after H2O2-Induced Cell InjuryMMP disorders and ROS overload are closely related to neuronal injury.37,38) To explore the roles of DNLA treatment on oxidative stress during H2O2-induced injury, MMP and ROS production were examined using JC-1 and DCFH-DA dyes, respectively. DNLA inhibited the fall of mitochondrial potential in cells treated with H2O2 compared with the control group (Figs. 3A, B). Similarly, DNLA treatment markedly reduced ROS production in cells during H2O2-induced injury (Figs. 3C, D), indicating that DNLA treatment inhibited oxidative stress responses during H2O2-induced cell injury.
DNLA Enhances Expression of Genes Encoding Brain Neuromarkers in N2A CellsBrain-derived neurotrophic factor (BDNF) and tyrosine hydroxylase (TH) play important roles in regulating homeostasis of neurotransmitter metabolism in the brain. The response of BDNF gene and TH were monitored by quantitative RT-PCR detection (Fig. 4). Cell treatment with 35 ng/mL DNLA alone showed a 2.61–2.24-fold increase in expression of BDNF and TH genes compared to controls, respectively. However, with 100 µM H2O2 treatment, hydrogen peroxide resulted in decreased gene expression. Interestingly, after treating DNLA with N2A cells, BDNF and TH increased by 5.04 and 4.27 times, respectively.
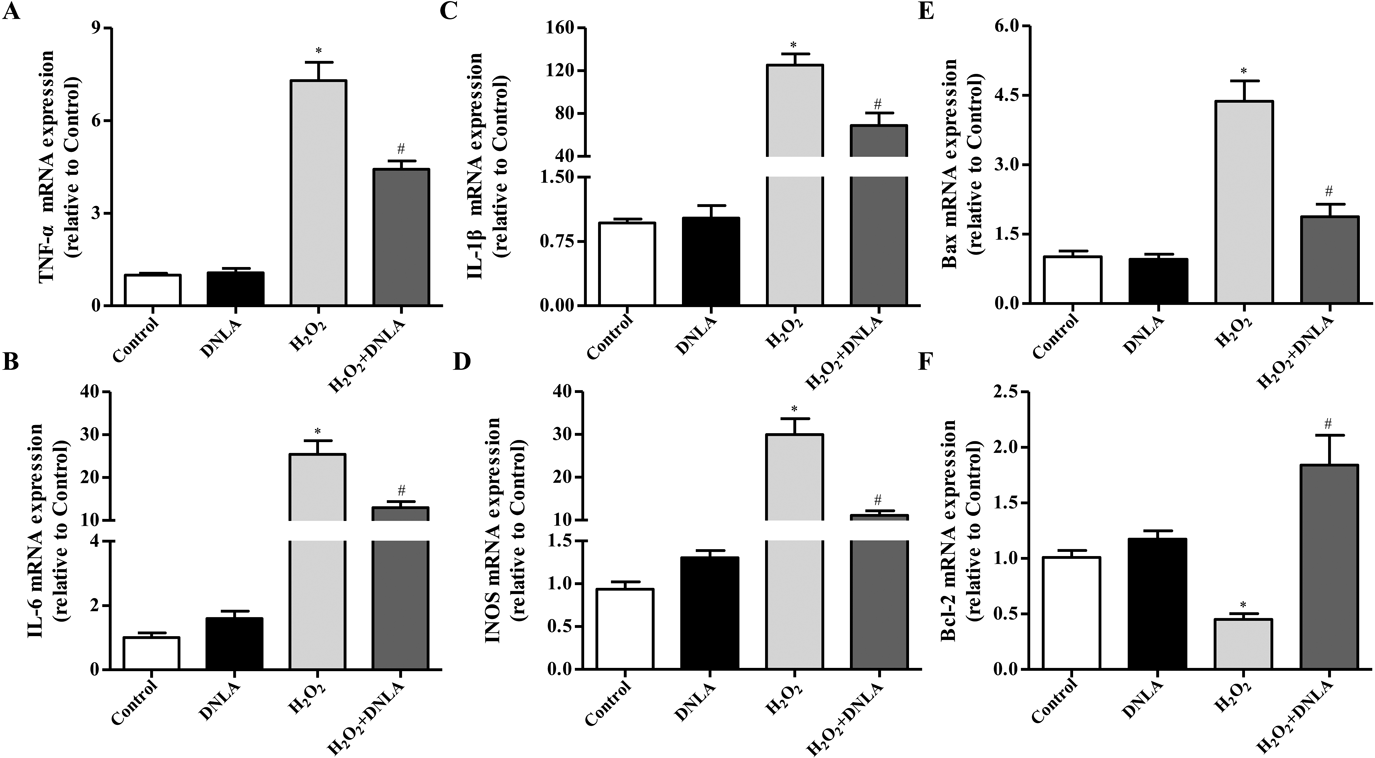
DNLA Suppression of H2O2-Induced Expression of Neuron pro-Inflammatory and Apoptotic FactorsTo explore the impact of DNLA on inflammation and apoptosis when exposed to H2O2, we used qPCR to determine inflammatory cytokines and apoptotic factors. Results showed that mRNA expression of inflammatory cytokines (tumor necrosis factor (TNF)-α, IL-6, IL-1β and iNOS) and cell apoptosis-related genes (Bax) were significantly elevated following H2O2 treatment. DNLA treatment significantly reduced TNF-α, IL-6, IL-1β, iNOS, and Bax mRNA expression, suggesting that DNLA markedly alleviates inflammatory and apoptotic responses (Figs. 5A–E). Bcl-2 is a well-known anti-apoptotic factor that binds to Bax. Our qPCR results showed that DNLA distinctly inhibited H2O2-induced increase in Bax and decrease in Bcl-2 mRNA expression (Figs. 5E, F).
Effect of DNLA on Activation of JAK/STATs Signaling Pathway in H2O2-Induced Neuronal DamageThe JAK–STAT signaling is related to cell apoptosis and affects inflammatory signaling cascades triggered by H2O2, interferon-γ (IFN-γ), and other cytokines.39,40) As DNLA affects mRNA expression of inflammatory cytokines and apoptotic factors, we investigated whether the inhibitory effect of DNLA on the expression of pro-inflammatory and pro-apoptosis mediators was mediated via JAK–STATs.41) DNLA inhibited the phosphorylation of STAT1 and STAT3 24 h after H2O2 stimulation (Figs. 6A, B). DNLA also significantly blocked the phosphorylation of JAK1 and JAK2 24 h after H2O2 challenge (Figs. 6C, D). Therefore, DNLA potently inhibited JAK1 and JAK2, subsequently downregulated phosphorylation of STAT1 and STAT3.
DISCUSSION
Neurological disorders are a leading cause of death and disability worldwide. Development of pathological oxidative stress injury participates in this disease. Currently, many studies have considered the H2O2-induced cell injury model as the model by which to elucidate mechanisms related to neuro-inflammation and apoptosis-linked cell injury in vitro and its treatment outcomes,42–46) because H2O2 simulate hypoxic environments, including, ROS production and loss of MMP in neuron. Protection of DNLA against ischemia-induced injury in vivo has been reported.26) While, the neuroprotective role behind the underlying mechanism of DNLA in neuron damage-induced cell injury remains unclear. The current study showed that DNLA protects neural cells against H2O2-induced demage and death, and attenuates the upregulation of intracellular Ca2+, ROS production, and inhibition mitochondrial depolarization in N2A cells during H2O2-induced cell injury, via inhibition of JAK–STATs pathways.
Evidently, oxidative stress is an important factor in secondary neuronal injury because it results in decreased cell viability, production of ROS, depolarization of mitochondrial, and expression of pro-apoptotic and pro-inflammatory proteins.47,48) Immoderate ROS may contribute to cerebral ischemia and neurodegenerative processes, resulting in dysregulation Ca2+ homeostasis and loss of MMP.49) Meanwhile, LDH release is increased in N2A cells exposed to H2O2. We observed that DNLA protected H2O2-induced N2A cells from cell injury and significantly reduced Ca2+ release (Fig. 2). Exposure of N2A cells to H2O2 increased ROS generation (Fig. 3C). DNLA was able to reduce H2O2-induced ROS generation and attenuate generation of LDH, a product of increased lipid peroxidation in N2A cells (Figs. 3A, 1D).
H2O2, a strong oxidizer, induces pro-inflammatory cytokines and cell apoptosis. When activated by H2O2, neuron, may induce large amounts of pro-inflammatory mediators and apoptotic factor, including NO, and prostaglandin E2 (PGE2), as well as cytokines such as IL-1β, IL-6, TNF-α, iNOS and Bax. These pro-inflammatory mediators and apoptotic factor lead to the emergence of disease. Overexpressed pro-inflammatory mediators and pro-apoptotic factor further exacerbate cell injury in many acute and chronic diseases, including arteriosclerosis, arthritis, infectious diseases, and cancer.50,51) Accordingly, materials or compounds that suppress these pro-inflammatory mediators and pro-apoptotic factor have been considered as potential neuroprotectant. Our results demonstrated that pro-inflammatory cytokines (TNF-α, IL-6, IL-1β and iNOS) and cell apoptosis genes (Bax) were induced by H2O2 stimulation in N2A cells (Figs. 5A–F). Thus, DNLA may exert anti-inflammatory and anti-apoptotic effects on H2O2-induced N2A cell injury. DNLA also inhibited mRNA expression of iNOS, TNF-α, IL-6, and Bax indicating that DNLA downregulated the expression of inflammatory and apoptotic genes at the transcription level.
To further investigate the mechanism of anti-inflammatory and anti-apoptosis effects of DNLA, we focused on the transcription factors, including JAKs, and STATs involved in regulating inflammatory and apoptotic genes. Mechanistically, H2O2 induces ROS to activate nuclear factor-kappaB (NF-κB) singaling pathway, an important transcription factor for iNOS and COX-2 expression in neuron. H2O2 also activates activator protein 1 (AP-1) which is stimulated by mitogen-activated protein kinases (MAPKs), including extracellular signal-regulated kinase (ERK)1/2, p38 MAPKs, and c-Jun N-terminal kinase (JNK), thus enhancing pro-inflammatory gene expression in nerve cells.52) However, DNLA did not influence the activation of NF-κB or AP-1 (Supplementary Fig. 1). Besides NF-κB and AP-1 signaling, JAK–STATs signaling is crucial for the expression of genes encoding inflammatory and apoptotic enzymes.53)
The JAK–STATs pathway is also important for cytokine activation signals in nerve injury. This suggests that the effects of inhibition of DNLA on H2O2 to stimulate inflammation and apoptosis may be attributed to the inhibition of JAK–STATs signaling. Therefore, we found that DNLA blocked phosphorylation of both JAK and STATs (Figs. 6A, B), and thus inhibited the transcriptional activation of the promoter of the target gene bound to the translocation nuclear. These results suggest that DNLA may inhibit H2O2-elevated release of pro-inflammatory and pro-apoptosis mediators by blocking the activation of STAT1 and STAT3.
In some neurological disorders, abnormal pathological reactions enhance JAK–STATs signaling.54) Aberrant activation of JAK–STATs signaling exert critical effects on neuron damage-induced apoptosis and death seen in N2A cells.55) We observed that DNLA treatment suppressed H2O2-induced upregulation of the phosphorylation activity of JAK1, JAK2, STAT1 and STAT3, indicating that DNLA mediates neuron damage progression in N2A cells by inhibiting cellular apoptosis, which is linked to JAK–STATs signaling. The current study provided novel mechanistic evidence indicating that DNLA attenuates H2O2-induced apoptosis and cytotoxicity via inhibition of JAK–STATs pathway. This study shows the potential of DNLA as a treatment for cell injury, including anti-inflammation, anti-cytotoxicity, anti-apoptosis, anti-oxidative stress, anti-endoplasmic stress, and mitochondrial protection. However, since N2A neuroblastoma cells which were neither of human origin nor primary culture cells were used in this study, further investigation involving in vitro experiments which come from primary culture cells and in vivo animal experiments to clarify the effects of DNLA on different H2O2-treated cells or organs, may be required for confirmation.
Acknowledgments
This study was supported in part by the Henan Science and Technology Research Program (for social development field, 2017) of Dendrobium huoshanensis fingerprint analysis and biological compound preparation research (172102310440), China Postdoctoral Science Foundation (2019M652826).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Xie Q, Zhao H, Li N, Su L, Xu X, Hong Z. Protective effects of Timosaponin BII on oxidative stress damage in PC12 cells based on metabolomic. Biomed. Chromatogr., 32, e4321 (2018).
- 2) Esposito MP, Nakazato RK, Pedroso ANV, Lima MEL, Figueiredo MA, Diniz AP, Kozovits AR, Domingos M. Oxidant-antioxidant balance and tolerance against oxidative stress in pioneer and non-pioneer tree species from the remaining Atlantic Forest. Sci. Total Environ., 625, 382–393 (2018).
- 3) Zhang L, Feng L, Jiang WD, Liu Y, Jiang J, Li SH, Tang L, Kuang SY, Zhou XQ. The impaired flesh quality by iron deficiency and excess is associated with increasing oxidative damage and decreasing antioxidant capacity in the muscle of young grass carp. Aquacult. Nutr., 22, 191–201 (2016).
- 4) Pu J, Tian G, Li B, Chen D, Yu B. Trace mineral overload induced hepatic oxidative damage and apoptosis in pigs with long-term high-level dietary mineral exposure. J. Agric. Food Chem., 64, 1841–1849 (2016).
- 5) Gautam DK, Misro MM, Chaki SP, Sehgal N. H2O2 at physiological concentrations modulates Leydig cell function inducing oxidative stress and apoptosis. Apoptosis, 11, 39–46 (2006).
- 6) Piao MJ, Ahn MJ, Kang KA, Ryu YS, Hyun YJ, Shilnikova K, Zhen AX, Jeong JW, Choi YH, Kang HK. Particulate matter 2.5 damages skin cells by inducing oxidative stress, subcellular organelle dysfunction, and apoptosis. Arch. Toxicol., 92, 2077–2091 (2018).
- 7) Lorenz R. A casuistic rationale for the treatment of spastic and myocloni in a childhood neurodegenerative disease: neuronal ceroid lipofuscinosis of the type Jansky–Bielschowsky. Neuroendocrinol. Lett., 23, 387–390 (2002).
- 8) Pedersen MO, Larsen A, Pedersen DS, Stoltenberg M, Penkowa M. Metallic gold reduces TNFalpha expression, oxidative DNA damage and pro-apoptotic signals after experimental brain injury. Brain Res., 1271, 103–113 (2009).
- 9) Seemann S, Lupp A. Administration of AMD3100 in endotoxemia is associated with pro-inflammatory, pro-oxidative, and pro-apoptotic effects in vivo. J. Biomed. Sci., 23, 68 (2016).
- 10) van Delft MF, Smith DP, Lahoud MH, Huang DC, Adams JM. Apoptosis and non-inflammatory phagocytosis can be induced by mitochondrial damage without caspases. Cell Death Differ., 17, 821–832 (2010).
- 11) Zhao H, He Y, Li S, Sun X, Wang Y, Shao Y, Hou Z, Xing M. Subchronic arsenism-induced oxidative stress and inflammation contribute to apoptosis through mitochondrial and death receptor dependent pathways in chicken immune organs. Oncotarget, 8, 40327–40344 (2017).
- 12) Chen SH, Lin JK, Liu SH, Liang YC, Lin-Shiau SY. Apoptosis of cultured astrocytes induced by the copper and neocuproine complex through oxidative stress and JNK activation. Toxicological Sciences an Official Journal of the Society of Toxicology, 102, 138–149 (2008).
- 13) Spanel-Borowski K. The ovulatory period as acute inflammatory response to oxidative stress and follicle cell harvest. Atlas of the Mammalian Ovary, 1, 39–59 (2012).
- 14) Magalhaes AC, Ashton FM. Effect of dicamba on oxygen uptake and cell membrane permeability in leaf tissue of cyperus rotundus L. Weed Res., 9, 48–52 (1969).
- 15) Mioka T, Fujimurakamada K, Mizugaki N, Kishimoto T, Sano T, Nunome H, Williams DE, Andersen RJ, Tanaka K. Phospholipid flippases and Sfk1p, a novel regulator of phospholipid asymmetry, contribute to low permeability of the plasma membrane. Mol. Biol. Cell, 29, 1203–1218 (2018).
- 16) Panahi G, Pasalar P, Zare M, Rizzuto R, Meshkani R. High glucose induces inflammatory responses in HepG2 cells via the oxidative stress-mediated activation of NF-κB, and MAPK pathways in HepG2 cells. Arch. Physiol. Biochem., 124, 1 (2018).
- 17) Lau WB, Ohashi K, Wang Y, Ogawa H, Murohara T, Ma X-L, Ouchi N. Role of adipokines in cardiovascular disease. Circ. J., 81, 920–928 (2017).
- 18) Yu J, Deng Y, Tao Z, Liang W, Guan X, Wu J, Ning X, Liu Y, Liu Q, He Z. The effects of HAP and macrophage cells to the expression of inflammatory factors and apoptosis in HK-2 cells of vitro co-cultured system. Urolithiasis, 46, 429–443 (2018).
- 19) Qi SM, Li Q, Jiang Q, Qi ZL, Zhang Y. Chrysin inhibits lipopolysaccharide-induced inflammatory responses of macrophages via JAK–STATs signaling pathway. Journal of Southern Medical University, 38, 243–250 (2018).
- 20) Yan Z, Gibson SA, Buckley JA, Qin H, Benveniste EN. Role of the JAK/STAT signaling pathway in regulation of innate immunity in neuroinflammatory diseases. Clinical Immunology, 189, 4–13 (2018).
- 21) Yang X, Mao X, Ding X, Guan F, Jia Y, Luo L, Li B, Tan H, Cao C. miR-146a down-regulation alleviates H2O2-induced cytotoxicity of PC12 cells by regulating MCL1/JAK/STAT pathway. Cell Biol. Toxicol., 34, 1–11 (2018).
- 22) Tyagi A, Agarwal C, Dwyer-Nield LD, Singh RP, Malkinson AM, Agarwal R. Silibinin modulates TNF-α and IFN-γ mediated signaling to regulate COX2 and iNOS expression in tumorigenic mouse lung epithelial LM2 cells. Mol. Carcinog., 51, 832–842 (2012).
- 23) Tsoyi K, Kim HJ, Shin JS, Kim DH, Cho HJ, Lee SS, Ahn SK, Yunchoi HS, Lee JH, Seo HG, Chang KC. HO-1 and JAK-2/STAT-1 signals are involved in preferential inhibition of iNOS over COX-2 gene expression by newly synthesized tetrahydroisoquinoline alkaloid, CKD712, in cells activated with lipopolysacchride. Cell. Signal., 20, 1839–1847 (2008).
- 24) Han HS, Jang E, Shin JS, Inn KS, Lee JH, Park G, Jang YP, Lee KT. Kyungheechunggan-Tang-01, a new herbal medication, suppresses LPS-induced inflammatory responses through JAK/STAT signaling pathway in RAW264.7 macrophages. Evid. Based Complement. Alternat. Med., 2017, 7383104 (2017).
- 25) Li Y, Li F, Gong Q, Wu Q, Shi J. Inhibitory effects of Dendrobium alkaloids on memory impairment induced by lipopolysaccharide in rats. Planta Med., 77, 117–121 (2011).
- 26) Zhang W, Wu Q, Lu YL, Gong QH, Zhang F, Shi JS. Protective effects of Dendrobium nobile Lindl. alkaloids on amyloid beta (25–35) -induced neuronal injury. Neural Regen. Res., 12, 1131–1136 (2017).
- 27) Xu YY, Xu YS, Wang Y, Wu Q, Lu YF, Liu J, Shi JS. Dendrobium nobile Lindl. alkaloids regulate metabolism gene expression in livers of mice. J. Pharm. Pharmacol., 69, 1409–1417 (2017).
- 28) Huang S, Wu Q, Liu H, Ling H, He Y, Wang C, Wang Z, Lu Y, Lu Y. Alkaloids of Dendrobium nobile Lindl. Altered hepatic lipid homeostasis via regulation of bile acids. J. Ethnopharmacol., 241, 111976 (2019).
- 29) Shu Y, Qihai G, Qin W, Fei L, Yuanfu L, Jingshan S. Alkaloids enriched extract from Dendrobium nobile Lindl. attenuates tau protein hyperphosphorylation and apoptosis induced by lipopolysaccharide in rat brain. Phytomedicine International Journal of Phytotherapy & Phytopharmacology, 21, 712–716 (2014).
- 30) Li S, Zhou J, Xu S, Li J, Liu J, Lu Y, Shi J, Zhou S, Wu Q. Induction of Nrf2 pathway by Dendrobium nobile Lindl. alkaloids protects against carbon tetrachloride induced acute liver injury. Biomed. Pharmacother., 117, 109073 (2019).
- 31) Zhang W, Wu Q, Lu YL, Gong QH, Shi JS. Protective effects of Dendrobium nobile Lindl. alkaloids on amyloid beta (25–35)-induced neuronal injury. Neural Regeneration Research, 12, 1131–1136 (2017).
- 32) Li LS, Lu YL, Nie J, Xu YY, Zhang W, Yang WJ, Gong QH, Lu YF, Lu Y, Shi JS. Dendrobium nobile Lindl alkaloid, a novel autophagy inducer, protects against axonal degeneration induced by Aβ25-35 in hippocampus neurons in vitro. CNS Neurosci. Ther., 23, 329–340 (2017).
- 33) Nam KN, Yae CG, Hong JW, Cho DH, Lee JH, Lee EH. Paeoniflorin, a monoterpene glycoside, attenuates lipopolysaccharide-induced neuronal injury and brain microglial inflammatory response. Biotechnol. Lett., 35, 1183–1189 (2013).
- 34) Fan LW, Mitchell HJ, Rhodes PG, Cai Z. α-Phenyl-n-tert-butyl-nitrone attenuates lipopolysaccharide-induced neuronal injury in the neonatal rat brain. Neuroscience, 151, 737–744 (2008).
- 35) Hua F, Ma J, Ha T, Kelley JL, Kao RL, Schweitzer JB, Kalbfleisch JH, Williams DL, Li C. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Res., 1262, 100–108 (2009).
- 36) Lu Y, Gu Y, Ding X, Wang J, Chen J, Miao C. Intracellular Ca2+ homeostasis and JAK1/STAT3 pathway are involved in the protective effect of propofol on BV2 microglia against hypoxia-induced inflammation and apoptosis. PLOS ONE, 12, e0178098 (2017).
- 37) Moon Y, Lee KH, Park JH, Geum D, Kim K. Mitochondrial membrane depolarization and the selective death of dopaminergic neurons by rotenone: protective effect of coenzyme Q10. J. Neurochem., 93, 1199–1208 (2005).
- 38) Nayak D, Kumari M, Rajachandar S, Ashe S, Thathapudi NC, Nayak B. Biofilm impeding AgNPs target skin carcinoma by inducing mitochondrial membrane depolarization mediated through ROS production. ACS Appl. Mater. Interfaces, 8, 28538-28553 (2016).
- 39) Murray PJ. The JAK–STAT signaling pathway: input and output integration. J. Immunol., 178, 2623–2629 (2007).
- 40) Badiei A, Gieseg S, Davies S, Izani OM, Bhatia M. LPS up-regulates cystathionine γ-lyase gene expression in primary human macrophages via NF-κB/ERK pathway. Inflamm. Allergy Drug Targets, 14, 99 (2015).
- 41) Okugawa S, Ota Y, Kitazawa T, Nakayama K, Yanagimoto S, Tsukada K, Kawada M, Kimura S. Janus kinase 2 is involved in lipopolysaccharide-induced activation of macrophages. Am. J. Physiol. Cell Physiol., 285, C399–C408 (2003).
- 42) Ma L, Guo X, Chen W. Inhibitory effects of oleoylethanolamide (OEA) on H2O2-induced human umbilical vein endothelial cell (HUVEC) injury and apolipoprotein E knockout (ApoE−/−) atherosclerotic mice. Int. J. Clin. Exp. Pathol., 8, 6301–6311 (2015).
- 43) Yin YL, Li P, Yang J, Liu DW, Sun RL, Pan GP, Wan GM, Wan GR. Protective effect of ultrafiltered XinMaiJia extract against H2O2-induced injury in human umbilical vein endothelial cells through NHE1 downregulation. Genet. Mol. Res., 13, 8436–8449 (2014).
- 44) Li R, Yin F, Guo YY, Zhao KC, Ruan Q, Qi YM. Knockdown of ANRIL aggravates H2O2-induced injury in PC-12 cells by targeting microRNA-125a. Biomed. Pharmacother., 92, 952–961 (2017).
- 45) Wang Z, Wu G, Yu Y, Liu H, Yang B, Kuang H, Wang Q. Xanthones isolated from Gentianella acuta and their protective effects against H2O2-induced myocardial cell injury. Nat. Prod. Res., 32, 2171–2177 (2018).
- 46) Zhang W, Zhu N, Hu M, Yu S, Sun Z, Wu H, Li P, Yang J, Ma G, Xu X. Congmujingnosides B-G, triterpene saponins from the stem of Aralia chinensis and their protective effects against H2O2-induced myocardial cell injury. Nat. Prod. Res., 33, 500-505 (2017).
- 47) Zhu Y, Liu Z, Peng YP, Qiu YH. Interleukin-10 inhibits neuroinflammation-mediated apoptosis of ventral mesencephalic neurons via JAK–STAT3 pathway. Int. Immunopharmacol., 50, 353–360 (2017).
- 48) Chen CX, Huang J, Tu GQ, Lu JT, Xie X, Zhao B, Wu M, Shi QJ, Fang SH, Wei EQ, Zhang WP, Lu YB. NAMPT inhibitor protects ischemic neuronal injury in rat brain via anti-neuroinflammation. Neuroscience, 356, 193–206 (2017).
- 49) Graham SH, Liu H. Life and death in the trash heap: the ubiquitin proteasome pathway and UCHL1 in brain aging, neurodegenerative disease and cerebral ischemia. Ageing Res. Rev., 34, 30–38 (2017).
- 50) Muniraj N, Stamp LK, Badiei A, Hegde A, Cameron V, Bhatia M. Hydrogen sulfide acts as a pro†inflammatory mediator in rheumatic disease. International Journal of Rheumatic Diseases, 20, 182–189 (2017).
- 51) Montacute R, Foley K, Forman R, Else KJ, Cruickshank SM, Allan MR. Enhanced susceptibility of triple transgenic Alzheimer’s disease (3xTg-AD) mice to acute infection. J. Neuroinflammation, 14, 50 (2017).
- 52) Lee SH, Na SI, Heo JS, Kim MH, Kim YH, Lee MY, Kim SH, Lee YJ, Han HJ. Arachidonic acid release by H2O2 mediated proliferation of mouse embryonic stem cells: involvement of Ca2+/PKC and MAPKs-induced EGFR transactivation. J. Cell. Biochem., 106, 787–797 (2009).
- 53) Yu H, Liu Z, Zhou H, Dai W, Chen S, Shu Y, Feng J. JAK–STAT pathway modulates the roles of iNOS and COX-2 in the cytoprotection of early phase of hydrogen peroxide preconditioning against apoptosis induced by oxidative stress. Neurosci. Lett., 529, 166–171 (2012).
- 54) Dominguez E, Mauborgne A, Mallet J, Desclaux M, Pohl M. SOCS3-mediated blockade of jak/stat3 signaling pathway reveals its major contribution to spinal cord neuroinflammation and mechanical allodynia after peripheral nerve injury. J. Neurosci., 30, 5754–5766 (2010).
- 55) Wang G, Qian P, Xu Z, Zhang J, Wang Y, Cheng S, Cai W, Qian G, Wang C, Decoster MA. Regulatory effects of the JAK3/STAT1 pathway on the release of secreted phospholipase A2-IIA in microvascular endothelial cells of the injured brain. J. Neuroinflammation, 9, 170–170 (2012).