Abstract
Fms-like tyrosine kinase 3 (FLT3) and isocitrate dehydrogenase 1/2 (IDH1/2) mutations drive malignancy in acute myeloid leukemia (AML), which accounts for approximately 40% of AML cases. Treatment with FLT3 or IDH1/2 inhibitors is used for such patients; however, it is not considered for most patients with AML who lack mutations on the respective genes. In this study, p90 ribosomal S6 kinase (RSK) was found to serve as a new therapeutic target in various AMLs with or without FLT3 mutations. BI-D1870, a potent inhibitor of RSK, significantly suppressed the proliferation of AML cell lines, among which three encoded wild-type FLT3 and three contained FLT3 driver mutations, compared with chronic myeloid leukemia K562 cells or other adherent cancer cells. BI-D1870 inhibited protein synthesis by dephosphorylating the p70 S6 kinase and eukaryotic initiation factor 4E-binding protein 1 in all AML cells except KG-1a cells. Meanwhile, the expression of microtubule-associated protein light chain 3B-I and -II increased in KG-1a cells treated with BI-D1870. BI-D1870 induced caspase-dependent apoptosis in all AML cells, including KG-1a cells. We next investigated the synergistic effect of BI-D1870 with cytarabine, a traditional anticancer drug used in AML. Synergistic effects of BI-D1870 and cytarabine were not observed in any of the cell lines. The findings suggested that BI-D1870 alone exerts an adequate antiproliferative effect on AML with or without FLT3 mutations and serves as a novel AML therapeutic agent.
INTRODUCTION
Acute myeloid leukemia (AML) is the most common type of acute leukemia; however, it has a poor diagnosis, with only 28.7% 5-year survival, as shown in a survey conducted between 2010 and 2016 in the U.S.A.1,2) The 5-year survival rate is over 60–70% in pediatric patients, whereas it is only 5–15% in elderly patients (>65 years old).2–5) AML is a genetically heterogeneous hematological malignancy in which the bone marrow cells are replaced by immature myeloid blasts generated by the growth of abnormal white blood cells. Molecular genetic analyses of AML indicated the gradual occurrence of genomic mutations during cell production from hematopoietic stem cells.6,7) Even though researchers have a thorough understanding of AML development, traditional anticancer treatments are typically prescribed in clinical settings. Massive dose therapy using cytarabine (Ara-C) is most commonly used in AML, and combination treatment with anthracyclines or hematopoietic stem cell transplantation has been implemented in the last few decades.
Recently, molecular targeted therapies using inhibitors against Fms-like tyrosine kinase 3 (FLT3) or isocitrate dehydrogenase (IDH) 1/2 have led to improved clinical outcomes in patients with AML. FLT3 inhibitors specifically inhibit the activity of mutant FLT3, which harbors mutations such as internal tandem duplication (ITD) and tyrosine kinase domain (TKD) alterations, and interrupt downstream signaling pathways involved in cell survival and proliferation, such as the phosphoinositide 3-kinase (PI3K)–AKT, mitogen-activated protein kinase (MAPK), and Janus kinase 2–signal transducer and activator of transcription 5 (STAT5) pathways.8–10) In contrast, IDH1 and IDH2 inhibitors inhibit the activity of IDH1 and IDH2 enzymes with point mutations, respectively, and suppress aberrant glycometabolism, thereby blocking differentiation and leukemia development.11) Although high remission rates have been reported for each drug in clinical trials, drug resistance has also been reported.12–16) In particular, several resistant FLT3 mutants were identified in patients treated with FLT3 inhibitors.12–15) Findings from our previous study suggested that heat shock protein 90 (HSP90) inhibitors may help counter resistance to FLT3 inhibitors by promoting rapid lysosomal degradation of mutant FLT3 proteins.17) Unfortunately, treatment with clinically usable HSP90 inhibitors has not been approved yet, and other chemotherapeutic strategies should be designed to overcome the FLT3 inhibitor resistance.
The MAPK signaling pathway is regulated by several tyrosine kinase receptors, and ligand binding prompts the gradual phosphorylation and activation of kinases, including the mitogenactivated protein kinase kinase (MEK)–extracellular signal-regulated kinase (ERK)–p90 ribosomal S6 kinase (RSK).18,19) RSKs (the four isoforms RSK1–4) are involved in multiple cellular events, including cell proliferation and survival. RSKs are expressed in normal cells, such as hematopoietic cells, in which they promote maturation and proliferation in response to the human cytokine granulocyte-macrophage colony-stimulating factor.20) In contrast, overexpression and/or hyperactivation of RSKs has been observed in various cancers, such as prostate, breast, lung, and kidney cancers, and blood tumors.21) RSK regulates the progression of solid tumors under stimulation with inflammatory cytokines and growth factors.22)
RSKs have been reported to activate mammalian target of rapamycin (mTOR) complex 1 (mTORC1) by phosphorylating regulatory associated protein of mTOR (Raptor), which is a component of mTORC1,23) and enhance the proliferation and survival of AML cells by phosphorylating ribosomal protein S6 (RPS6).24) PI3K–AKT signaling regulates mTORC1 expression by inhibiting the expression of the negative regulator Tuberin/tuberous sclerosis 2.25) To promote protein synthesis, activated mTORC1 first phosphorylates the ribosomal protein S6 kinase beta-1, also known as p70 S6 kinase (p70S6K), on Thr389, then phosphorylates RPS6, and finally induces protein translation by phosphorylating eukaryotic initiation factor 4E-binding protein 1 (4E-BP1) at Thr37/46.26,27)
BI-D1870 is a derivative of a novel series of dihydropteridinones that acts as a cell-permeable ATP-competitive inhibitor of RSK isoforms.28) It has multiple anticancer attributes, such as reactive oxygen species generation, and increases endoplasmic reticulum (ER) stress and autophagy.29) In our previous study, BI-D1870 lowered the stability of P-glycoprotein and helped overcame resistance to anticancer drugs.30) BI-D1870 exhibits potential anticancer effects against multiple tumor types, including AML, via various mechanisms. However, its cytotoxicity has also been suggested, as RSKs contribute to the cytokine-mediated proliferation of hematopoietic cells.
Approximately 30% of patients with AML carry FLT3 mutations, including ITDs and TKD alterations, and only 5–10% of patients carry IDH1 or IDH2 mutations.10) More than 50% of patients with AML carry no mutations in FLT3 or IDH1/2; for these patients, molecular targeted drugs that are currently approved for AML treatment are not used. In such cases, patients receive only standard therapy with traditional anticancer agents, including Ara-C. However, the remission induction rates are almost never high; therefore, other treatment methods should be considered, alongside the development of novel therapies with molecular targeted drugs.
In this study, we demonstrated that BI-D1870 suppresses the proliferation of AML cells with or without FLT3-ITD mutations. The drug inhibited protein synthesis by downregulating the phosphorylation of 4E-BP1 and p70S6K in AML cell lines, except KG-1a cells, and induced apoptosis in all AML cell lines. Collectively, RSK could act as a molecular target in AML treatment regardless of the presence or absence of mutations such as FLT3-ITD.
MATERIALS AND METHODS
ReagentsPuromycin and 3-(4,5-dimethylthial-2-yl)-2,5-diphenyltetrazalium bromide (MTT) were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Bafilomycin A1 and Ara-C were obtained from MedChemExpress (Monmouth Junction, NJ, U.S.A.) and Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan), respectively. Other inhibitors were purchased from Selleck Chemicals LLC (Houston, TX, U.S.A.).
CellsMV4-11 cells were purchased from the American Type Culture Collection (Manassas, VA, U.S.A.) and cultured in Iscove’s modified Dulbecco’s medium (FUJIFILM Wako) supplemented with 10% fetal bovine serum (FBS), 100 units/mL penicillin G, and 100 units/mL streptomycin (FUJIFILM Wako). Kasumi-1 cells were obtained from the JCRB Cell Bank, National Institute of Biomedical Innovation (Osaka, Japan), and maintained in RPMI-1640 medium (FUJIFILM Wako) supplemented with 20% FBS and penicillin-streptomycin. MOLM-13 and MOLM-14 cells were obtained from JCRB Cell Bank, KG-1a cells were obtained from RIKEN BRC (Tsukuba, Japan), and HL60 and K562 cells were obtained from NIH-NCI (Bethesda, MD, U.S.A.). These cells were cultured in RPMI-1640 medium supplemented with 10% FBS and antibiotics. All cells were cultured at 37 °C in a humidified atmosphere with 5% CO2.
Growth Inhibition AssayKasumi-1 or other types of cells were seeded on a 96-well plate at 1 × 105 or 3 × 104 cells/100 µL/well, respectively, and treated with 50 µL of increasing concentrations of inhibitors. After a 4-d culture period, the MTT assay was performed as described previously.27,28) The number of cells surviving after treatment with the inhibitors was determined using a Coulter Counter Z1 (Beckman Coulter Inc., Brea, CA, U.S.A.). The IC50 values were determined from the growth inhibition curves, as described previously27,28); data are presented as mean ± standard deviation (S.D.) from triplicate experiments.
ImmunoblottingImmunoblotting was performed as described previously.30–32) Proteins were resolved using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), electrotransferred to an Immobilon-P membrane (EMD Millipore, Billerica, MA, U.S.A.), treated with primary antibodies, and then treated with peroxidase-conjugated anti-rabbit or anti-mouse secondary antibody (Cell Signaling Technology, Inc., Danvers, MA, U.S.A.). An anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (6C5; Merck Millipore, Burlington, MA, U.S.A.), anti-puromycin antibody (3RH11; Cosmo Bio Co., Ltd., Tokyo, Japan), and other antibodies (Cell Signaling Technology, Inc.) were used for immunoblotting.
Apoptosis DNA Ladder AssayThe cultured cells were collected with the culture media and centrifuged at 200 × g for 5 min at 4 °C. After the supernatant was discarded, the cells were suspended in 500 µL of detergent buffer (10 mM Tris–HCl [pH 7.5], 5 mM ethylenediaminetetraacetic acid (EDTA) [pH 7.5], and 0.2% Triton X-100), incubated on ice for 30 min, and centrifuged at 20000 × g for 45 min at 4 °C. The supernatant was collected in a new tube; 100 µL of 5 M NaCl, 1.2 mL of ethanol, and 300 µL of 3 M sodium acetate (pH 5.2) were added, and the solution was incubated for 1 h at −80 °C. After centrifugation at 20000 × g for 20 min at 4 °C, the supernatant was discarded, and the DNA extracts were dissolved in 400 µL of TE buffer (10 mM Tris–HCl [pH 8.0] and 5 mM EDTA [pH 8.0]) and 2 µL of 10 mg/mL deoxyribonuclease (DNase)-free ribonuclease (RNase) (Nippongene, Tokyo, Japan) and incubated for 5 h at 37 °C. Twenty-five microliters of 20 mg/mL proteinase K (FUJIFILM Wako) and 40 µL of a buffer (10 mM Tris–HCl [pH 8.0], 5 mM EDTA [pH 8.0], and 500 mM NaCl) were added, and the sample was incubated overnight at 65 °C. After cooling to room temperature, 467 µL of chloroform-isoamyl alcohol (25 : 24 : 1) (FUJIFILM Wako) was added, and the tube was centrifuged at 12000 × g for 2 min at 4 °C. The aqueous phase (top layer) was transferred to a fresh tube. Three volumes of ethanol and one-tenth volume of 3 M sodium acetate (pH 5.2) were added, and the tube was incubated for 1 h at −80 °C. After centrifugation at 20000 × g for 20 min at 4 °C, the supernatant was discarded, the pellet was air-dried, and DNA was dissolved in 20 µL of TE buffer. The extracted DNA was quantified using a spectrophotometer NanoDrop 1000 (Thermo Fisher Scientific, Waltham, MA, U.S.A.). The DNA samples (1 µg) were mixed with Midori Green Direct (NIPPON Genetics Co., Ltd., Tokyo, Japan) and electrophoresed on a 2% agarose gel (FUJIFILM Wako). The gel was photographed using an LED transilluminator at 470 nm.
RESULTS
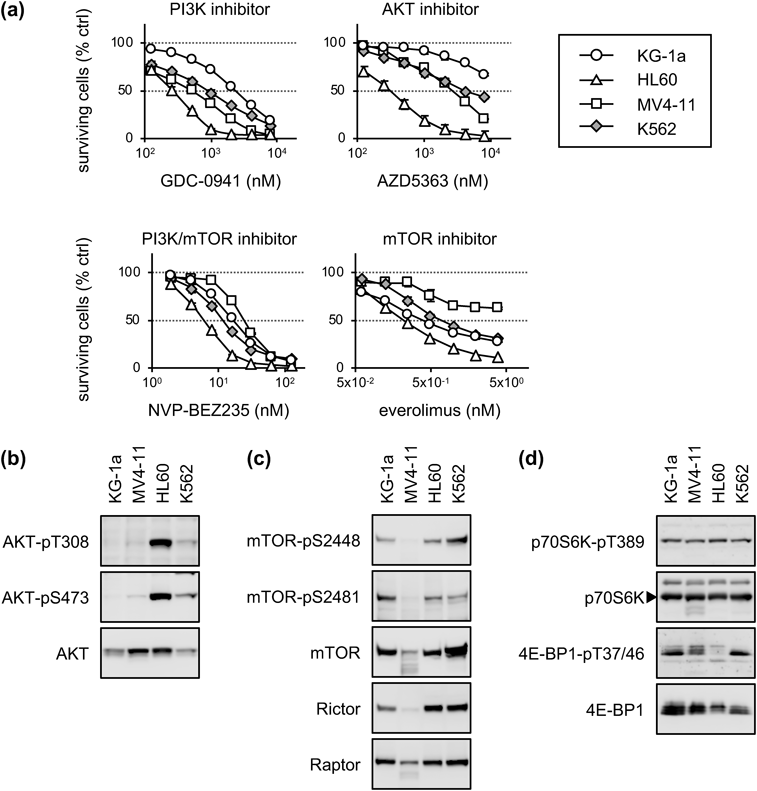
p70S6K and 4B-BP1 Were Expressed Ubiquitously in AML CellsFLT3 inhibitors are effective in cases of AML with FLT3 gene mutations, such as FLT3-ITD and/or FLT3-TKD; however, molecular targeted drugs for FLT3-WT AML are limited, although IDH1/2 inhibitors and the BCL2 inhibitor venetoclax have been approved. Therefore, we attempted to identify effective inhibitors of FLT3-WT AML cells; we had previously screened drugs with growth inhibitory activity in the cell lines.33) The screening revealed that NVP-BEZ235, a PI3K-mTOR dual inhibitor, showed higher growth inhibitory activity in HL60 cells, an FLT3-WT AML cell line, than in the chronic myeloid leukemia cell line K562. In addition, the mTOR inhibitors everolimus and rapamycin also showed higher growth inhibitory activity in HL60 and KG-1a cells than in K562 cells. To evaluate these results in detail, growth inhibition assays were performed for the FLT3-WT AML cell lines KG-1a and HL60, the FLT3-ITD cell line MV4-11, and K562 cells using GDC-0941 (a PI3K inhibitor), AZD5363 (an AKT inhibitor), NVP-BEZ235, and everolimus (Fig. 1a). Compared to K562 cells, HL60 cells showed greater sensitivity to all four inhibitors, and other AML cells showed similar or less sensitivity to the drugs. The sensitivity of AML cells to trametinib (an MEK inhibitor) or pimozide (a STAT5 inhibitor) was similar to that of K562 cells (Supplementary Fig. S1). To confirm the difference in drug sensitivity among the AML cell lines, the expression of proteins associated with growth signaling pathways, such as the AKT–mTORC1, MAPK, and STAT5 pathways, were evaluated using immunoblotting (Figs. 1b–d). Phosphorylated AKT was clearly detected in HL60 cells, whereas it was detected at low levels in the other cell lines (Fig. 1b). mTOR, Raptor, and Rictor, which are components of mTORC1 or mTORC2, were expressed at high levels in KG-1a, HL60, and K562 cells but at low levels in MV4-11 cells (Fig. 1c). All cells expressed similar levels of p70S6K and 4E-BP1 (Fig. 1d). MV4-11 and K562 cells expressed phosphorylated ERK at high levels, whereas K562 cells expressed phosphorylated STAT5 at high levels (Supplementary Fig. S2). These results suggest that the proliferation of HL60 cells depends on the PI3K–AKT pathway, that of MV4-11 cells on the MAPK pathway, and that of K562 cells on the STAT5 pathway. In contrast, the addictive pathways of KG-1a growth are unclear. Thus, the proliferation of each cell line may depend on the different signaling pathways, and these signal inhibitors would not be appropriate for treatment of all cases of AML. However, the results suggested that the protein synthesis pathway mTORC1–p70S6K and the protein initiation pathway mTORC1–4E-BP1 are common pathways in the growth of these cells.
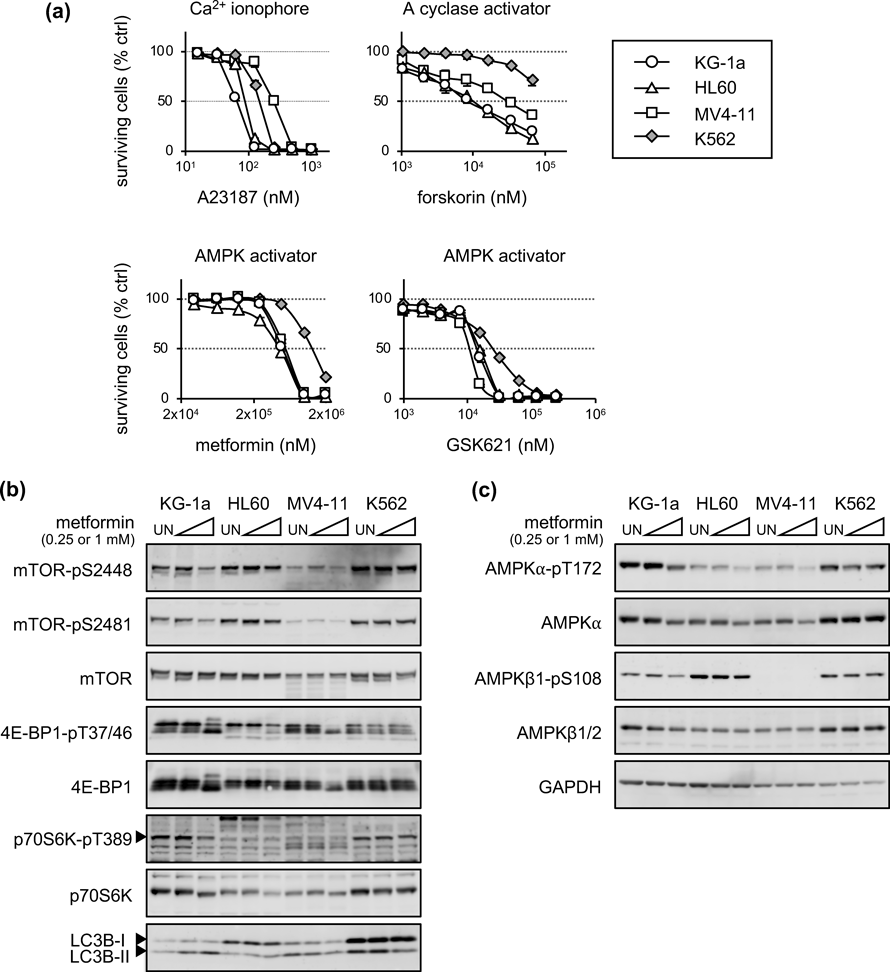
AMP-Activated Protein Kinase (AMPK) Activation Marginally Downregulated p70S6K and 4E-BP1AMPK signaling is known to negatively regulate mTORC1. A clinical trial on metformin (an AMPK activator) with Ara-C for AML treatment was performed in the U.S.A.34) In the present study, we examined the growth inhibitory activity of metformin and its related compounds (Fig. 2a). Compared to K562 cells, all AML cells showed greater sensitivity to forskorin (a cyclase activator), metformin, and GSK621 (an AMPK activator). KG-1a and HL60 cells were more sensitive to A23187 (Ca2+ ionophore) than K562 cells, whereas MV4-11 cells were more resistant. Next, the levels of mTOR (pS2448 and pS2481), 4E-BP1 (pT37/46), and p70S6K (pT389) phosphorylation after metformin treatment were evaluated using immunoblotting (Fig. 2b). A slight downregulation of phosphorylated mTOR (pS2448) and 4E-BP1 (pT37/46) was observed in KG-1a and MV4-11 cells treated with 1 mM metformin for 24 h, but not in HL60 and K562 cells. We assessed the phosphorylation of AMPK in cells treated with metformin using the same lysates (Fig. 2c). The level of phosphorylation at T172 in AMPKα was slightly lowered in all AML cells treated with metformin at a concentration of 1 mM, whereas the level of phosphorylation in AMPKβ (pS108) was not. These results indicated that AMPK activation inhibits AML cell growth, but the inhibitory effects on mTORC1 activity differ in each cell line. In addition, the effective concentrations of AMPK activators used in this study were considerably higher than the blood concentrations in clinical settings (approximately 5 µM). Therefore, we did not further evaluate the role of AMPK signaling in AML proliferation and instead assessed other signal inhibitors that suppress p70S6K and/or 4E-BP1 activity in the different AML cell lines.
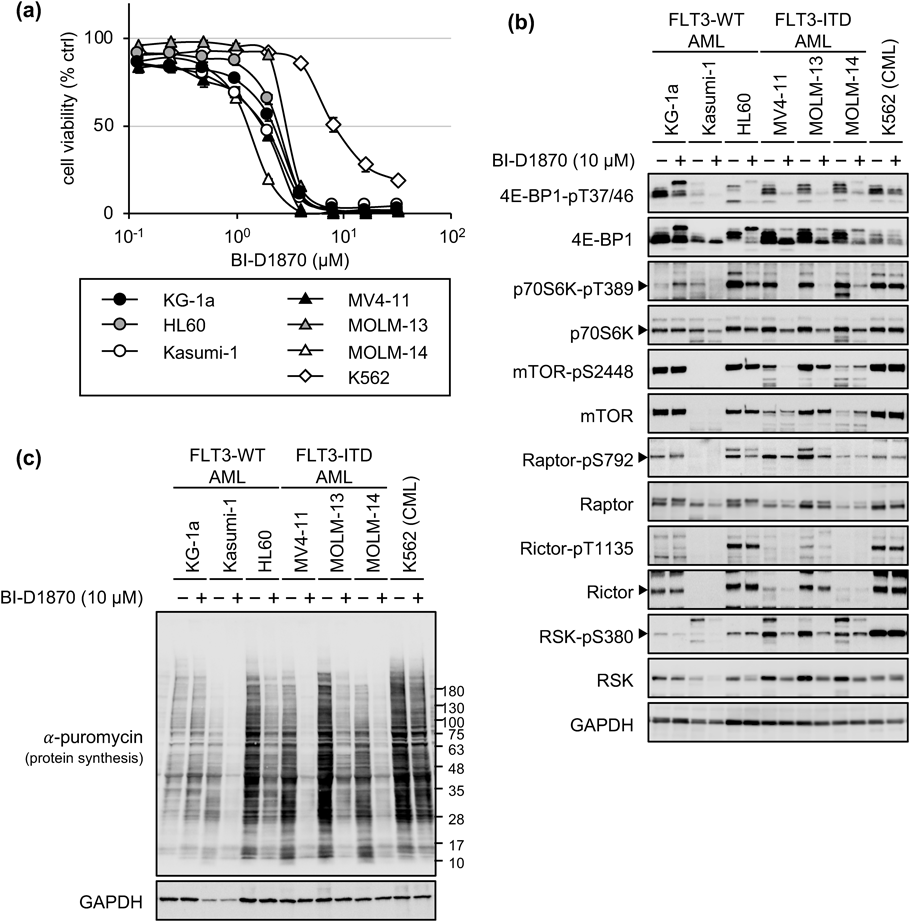
RSK Inhibition Suppressed AML Cell Proliferation by Downregulating Protein SynthesisNext, we investigated the signaling pathway related to mTORC1 in the growth inhibition of AML cells. Although several candidate inhibitors were identified in growth inhibition assays and immunoblotting experiments (data not shown), the pan-RSK inhibitor BI-D1870 was the most effective in suppressing AML cell proliferation. Surprisingly, compared to K562 and adherent cells, both FLT3-WT AML cells (KG-1a, HL60, and Kasumi-1) and FLT3-ITD cells (MV4-11, MOLM-13, and MOLM-14) were significantly more sensitive to BI-D1870 (Fig. 3a, Supplementary Figs. S3, S4; Table 1). We next treated the six types of AML cells and K562 cells with BI-D1870 for 24 h and analyzed the phosphorylation of 4E-BP1 and p70S6K (Fig. 3b). BI-D1870 downregulated the phosphorylation of 4E-BP1 (T37/46) and p70S6K (T389) in all AML cells, except KG-1a cells, but not in K562 cells. The levels of phosphorylation of Raptor (S792) decreased marginally in the AML cells. The levels of phosphorylation of mTOR (S2448) were also lowered in MV4-11 and MOLM-13 cells, but not in other cell lines. To analyze protein synthesis under BI-D1870 treatment, we made use of the previously determined concentration and incubation time of puromycin (1 µM and 1 h, respectively) before cell harvest (Supplementary Fig. S5). Consistent with the immunoblotting experiment, the puromycin uptake experiment (Fig. 3c) revealed that new protein synthesis was downregulated in response to BI-D1870 treatment in all AML cells except KG-1a cells (Fig. 3b). These results indicated that BI-D1870 suppresses the proliferation of AML cells by inhibiting protein synthesis and translation in almost all AML cell lines irrespective of the presence or absence FLT3 mutations.
Table 1. Cell Growth Inhibition Profile for BI-D1870
| Cell line | IC50 values (µM) | Sensitivity relative to that of K562 cells |
|---|
| KG-1a | 2.25 ± 0.12 | 0.27 |
| HL60 | 2.55 ± 0.08 | 0.30 |
| Kasumi-1 | 1.82 ± 0.12 | 0.21 |
| MV4-11 | 1.74 ± 0.11 | 0.20 |
| MOLM-13 | 3.15 ± 0.02 | 0.37 |
| MOLM-14 | 1.34 ± 0.02 | 0.16 |
| K562 | 8.49 ± 0.84 | — |
| HCT-15 | 20.07 ± 0.44 | 2.36 |
| HT-29 | 10.77 ± 0.53 | 1.27 |
| HCT 116 | 6.33 ± 0.07 | 0.75 |
| HT1080 | 23.31 ± 3.44 | 2.75 |
| KB-3-1 | 1.65 ± 0.17 | 0.19 |
IC50 values are the mean ± S.D. values obtained from three independent experiments. The relative sensitivity values were calculated by dividing the IC50 value for each cell line by the IC50 value of K562 cells.
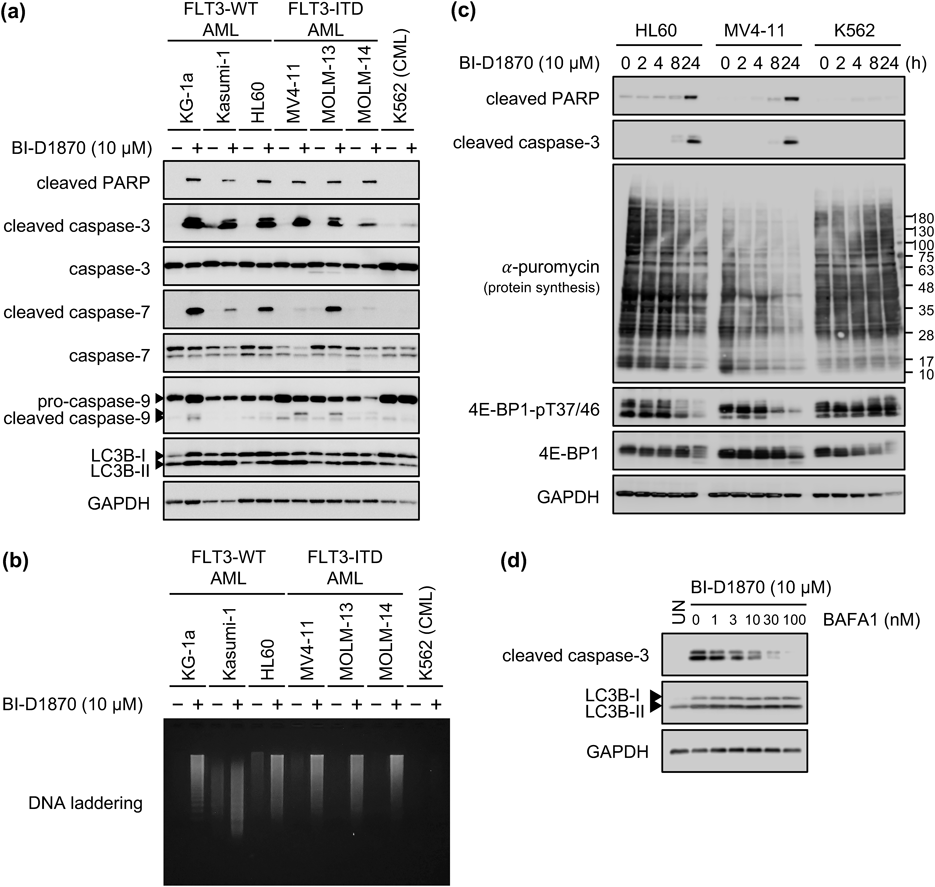
We investigated the induction of apoptosis via caspase activation. Results from the immunoblotting experiment (Fig. 4a) revealed that treatment with BI-D1870 induced the cleavage of poly(ADP-ribose)polymerase (PARP) and caspase-3 in all AML cell lines, but not in K562 cells. Cleaved caspase-7 was also detected in KG-1a, Kasumi-1, HL60, and MOLM-13 cells, and cleaved caspase-9 was only detected in KG-1a, MV4-11, and MOLM-13 cells. Under the same conditions used in the immunoblotting experiment, DNA laddering was observed only in AML cell lines upon treatment with BI-D1870 for 24 h (Fig. 4b). To investigate whether the downregulation of protein synthesis induced apoptosis or was only a result of cell death, time-course experiments (similar to those described in Fig. 4a) were performed in HL60, MV4-11, and K562 cells (Fig. 4c). After treatment with BI-D1870 for 8 h, protein synthesis and phosphorylated 4E-BP1 expression were downregulated in HL60 and MV4-11 cells, but not in K562 cells. In contrast, the cleavage of PARP and caspase-3 was observed in HL60 and MV4-11 cells treated with the drug for 24 h. These results indicated that BI-D1870-induced caspase-dependent apoptosis is mediated by the downregulation of protein synthesis. Next, we investigated the reason why BI-D1870 induced apoptosis without downregulating protein synthesis in KG-1a cells (Fig. 4d). Several groups have reported autophagic cell death35,36); here, we evaluated the effect of bafilomycin A1, a lysosome inhibitor, on BI-D1870-induced cell death in KG-1a cells. Bafilomycin A1 suppressed the cleavage of caspase-3 in a concentration-dependent manner, suggesting the possibility that the drug induced autophagy followed by apoptosis in KG-1a cells. These results indicated that BI-D1870 preferentially induces apoptosis in various AML cells than in other cell lines.
As Ara-C is used for standard AML therapy, we evaluated the synergistic effect of BI-D1870 and Ara-C in growth inhibition assays (Supplementary Fig. S6). Neither synergistic nor additive effects were observed in KG-1a, HL60, MOLM-13, and MOLM-14 cells, although the effect of each drug was clearly observed. In fact, at certain concentrations, Ara-C suppressed the effect of BI-D1870 on cell proliferation.
DISCUSSION
To explore AML therapies based on novel molecular targets that can be used for patients with or without specific genetic mutations, including FLT3 and/or IDH1/2 mutations associated with AML malignancy, we previously screened different inhibitors and found mTOR inhibitors to be ideal candidates.33) In this study, mTOR inhibitors and related compounds were evaluated in growth inhibition assays using several AML cell lines, and HL60 cells were found to be more sensitive to the inhibitors than K562 cells (Fig. 1). Meanwhile, KG-1a and MV4-11 cells were similar or less sensitive than K562 cells. We further attempted to identify inhibitors that suppress the proliferation of AML cells, with a focus on the regulators of mTORC1. BI-D1870 specifically suppressed the proliferation of AML cells via protein synthesis inhibition by downregulating the phosphorylation of 4E-BP1 and p70S6K, which are regulated by mTORC1, in all AML cell lines, except KG-1a cells (Fig. 3), and induced apoptosis in all AML cell lines (Fig. 4). Although BI-D1870 did not downregulate protein synthesis in KG-1a cells, it specifically increased the expression of microtubule-associated protein light chain 3 I and II (Figs. 4a, b), and bafilomycin A1 suppressed the cleavage of caspase-3 induced by BI-D1870 (Fig. 4b). These results suggested the possibility that BI-D1870 induces autophagy followed by apoptosis in KG-1a cells. To evaluate autophagic cell death in KG-1a cells treated with BI-D1870, transmission electron microscopy is required in the presence or absence of autophagy inhibitors, and this will be examined in future studies.
mTORC1 is a multiprotein assembly composed of Raptor, mTOR, Dep-domain mTOR interacting protein, and G protein beta subunit like. BI-D1870 marginally lowered the phosphorylation of the S792 site in Raptor, which conserves the phosphorylation recognition sequence of RSK, in all AML cell lines except KG-1a cells (Fig. 3b). This result indicated that BI-D1870 downregulated mTORC1 activity, which is consistent with the findings reported by Carrière et al.23) However, the extent of downregulation of Raptor phosphorylation was lower than that of the downregulation of 4E-BP1 and p70S6K phosphorylation. This suggested that the suppression of protein synthesis by RSK inhibition is partly mediated by mTORC1, but primarily by other molecules. The RSK-mediated direct phosphorylation of 4E-BP1 or p70S6K has never been reported, and the brief consensus sequences recognized and phosphorylated by RSK have not been identified in 4E-BP1 or p70S6K. Therefore, other unknown kinase(s) may be associated with the RSK-mediated phosphorylation of 4E-BP1 and p70S6K. RSKs phosphorylate S235/236, which are also the phosphorylation sites for p70S6K, in RPS6.37,38) Therefore, RSK inhibition possibly suppresses protein synthesis through the direct inhibition of RPS6.
Chae et al. reported that the levels of phosphorylated RSK in AML cells were higher than those in normal CD34-positive bone marrow samples, and that BI-D1870 inhibited AML proliferation by inducing mitotic arrest in HL60 cells.39) The authors reported that the molecular mechanism underlying BI-D1870-mediated cell cycle arrest involves the failure of CDC20 dissociation from MAD2. This finding supports our results showing BI-D1870-induced growth arrest and apoptosis in HL60 cells. In addition, we analyzed the effects of BI-D1870 in expanded AML cell lines that carried several gene mutations associated with AML malignancy and demonstrated that BI-D1870 is widely effective against multiple AML cell lines. Hence, RSK could serve as a promising target for AML treatment, and RSK inhibitors should be developed in the near future for use in clinical settings.
Acknowledgments
This work was, in part, supported by the Nihon University Chairman of the Board of Trustees Grant.
Author Contributions
KK designed the study, performed the experiments and data analyses, supervised the data, and wrote the manuscript. AN performed the experiments and analyzed the data. All authors approved and agreed to the final manuscript.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) NCI. NIH. “Cancer stat facts: leukemia — acute myeloid leukemia (AML).”: ‹https://seer.cancer.gov/statfacts/html/amyl.html›, accessed 17 February, 2021.
- 2) Noone AM, Howlader N, Krapcho M, Miller D, Brest A, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA. “SEER Cancer Statistics Review (CSR) 1975–2015.” National Cancer Institute. Bethesda, MD.: ‹https://seer.cancer.gov/csr/1975_2015/›, accessed 2 March, 2021.
- 3) Saygin C, Carraway HE. Emerging therapies for acute myeloid leukemia. J. Hematol. Oncol., 10, 93 (2017).
- 4) Tsukimoto I, Tawa A, Horibe K, Tabuchi K, Kigasawa H, Tsuchida M, Yabe H, Nakayama H, Kudo K, Kobayashi R, Hamamoto K, Imaizumi M, Morimoto A, Tsuchiya S, Hanada R. Risk-stratified therapy and the intensive use of cytarabine improves the outcome in childhood acute myeloid leukemia: the AML99 trial from the Japanese childhood AML cooperative study group. J. Clin. Oncol., 27, 4007–4013 (2009).
- 5) Miyamoto K, Minami Y. Precision medicine and novel molecular target therapies in acute myeloid leukemia: the background of hematologic malignancies (HM)-SCREEN-Japan 01. Int. J. Clin. Oncol., 24, 893–898 (2019).
- 6) Yang L, Rau R, Goodell MA. DNMT3A in hematological malignancies. Nat. Rev. Cancer, 15, 152–165 (2015).
- 7) Yang L, Rodriguez B, Mayle A, Park HJ, Lin X, Luo M, Jeong M, Curry CV, Kim SB, Ruau D, Zhang X, Zhou T, Zhou M, Rebel VI, Challen GA, Göttgens B, Lee JS, Rau R, Li W, Goodell MA. DNMT3A loss drives enhancer hypomethylation in FLT3-ITD-associated leukemias. Cancer Cell, 29, 922–934 (2016).
- 8) Larrosa-Garcia M, Baer MR. FLT3 inhibitors in acute myeloid leukemia: current status and future directions. Mol. Cancer Ther., 16, 991–1001 (2017).
- 9) Meshinchi S, Appelbaum FR. Structure and functional alterations of FLT3 in acute myeloid leukemia. Clin. Cancer Res., 15, 4263–4269 (2009).
- 10) Kihara R, Nagata Y, Kiyoi H, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia, 28, 1586–1595 (2014).
- 11) Martelli MP, Martino G, Cardinali V, Falini B, Martinelli G, Cerchione C. Enasidenib and ivosidenib in AML. Minerva Med., 111, 411–426 (2020).
- 12) Man CH, Fung TK, Ho C, Han HHC, Chow HCH, Ma ACH, Choi WWL, Lok S, Cheung AMS, Eaves C, Kwong YL, Leung AYH. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: favorable initial outcome and mechanisms of subsequent noresponsiveness associated with the emergence of a D835 mutation. Blood, 119, 5133–5143 (2012).
- 13) Smith CC, Lasater EA, Zhu X, Lin KC, Stewart WK, Damon LE, Salerno S, Shah NP. Activity of ponatinib against clinically-relevant AC220-resistant kinase domain mutants of FLT3-ITD. Blood, 121, 3165–3171 (2013).
- 14) Lin J, Zhang Y, Matusow B, Mumy A, Tsang G, Zhang J, Powers H, Spevak W, Severson P, Tsai J, Bollag G, Zhang C. A mixed type 1 and type 2 kinase inhibitor that overrides FLT3 F691 and D835 resistance mutations. Blood, 128, 1074 (2016).
- 15) Alvarado Y, Kantarjian HM, Luthra R, Ravandi F, Borthakur G, Garcia-Manero G, Konopleva M, Estrov Z, Andreeff M, Cortes JE. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer, 120, 2142–2149 (2014).
- 16) Intlekofer AM, Shih AH, Wang B, et al. Acquired resistance to IDH inhibition through trans or cis dimer-interface mutations. Nature, 559, 125–129 (2018).
- 17) Katayama K, Noguchi K, Sugimoto Y. Heat shock protein 90 inhibitors overcome the resistance to Fms-like tyrosine kinase 3 inhibitors in acute myeloid leukemia. Oncotarget, 9, 34240–34258 (2018).
- 18) Traverse S, Cohen P, Paterson H, Marshall C, Rapp U, Grand RJ. Specific association of activated MAP kinase kinase kinase (Raf) with the plasma-membranes of Ras-transformed retinal cells. Oncogene, 8, 3175–3181 (1993).
- 19) Chang F, Steelman LS, Lee JT, Shelton JG, Navolanic PM, Blalock WL, Franklin RA, McCubrey JA. Signal transduction mediated by the Ras/Raf/MEK/ERK pathway from cytokine receptors to transcription factors: potential targeting for therapeutic intervention. Leukemia, 17, 1263–1293 (2003).
- 20) Joseph DE, Paul CC, Baumann MA, Gomez-Cambronero J. S6 kinase p90rsk in granulocyte-macrophage colony-stimulating factor-stimulated proliferative and mature hematopoietic cells. J. Biol. Chem., 271, 13088–13093 (1996).
- 21) Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol., 9, 747–758 (2008).
- 22) Zhou Y, Sakurai H. Emerging and diverse functions of the EphA2 noncanonical pathway in cancer progression. Biol. Pharm. Bull., 40, 1616–1624 (2017).
- 23) Carrière A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, Roux PP. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol., 18, 1269–1277 (2008).
- 24) Watanabe D, Nogami A, Okada K, Akiyama H, Umezawa Y, Miura O. FLT3-ITD activates RSK1 to enhance proliferation and survival of AML cells by activating mTORC1 and eIF4B cooperatively with PIM or PI3K and by inhibiting Bad and BIM. Cancers (Basel), 11, 1827 (2019).
- 25) Dibble CC, Cantley LC. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol., 25, 545–555 (2015).
- 26) Ruvinsky I, Meyuhas O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci., 31, 342–348 (2006).
- 27) Hara K, Yonezawa K, Kozlowski MT, Sugimoto T, Andrabi K, Weng QP, Kasuga M, Nishimoto I, Avruch J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J. Biol. Chem., 272, 26457–26463 (1997).
- 28) Sapkota GP, Cummings L, Newell FS, Armstrong C, Bain J, Frodin M, Grauert M, Hoffmann M, Schnapp G, Steegmaier M, Cohen P, Alessi DR. BI-D1870 is a specific inhibitor of the p90 RSK (ribosomal S6 kinase) isoforms in vitro and in vivo. Biochem. J., 401, 29–38 (2007).
- 29) Chiu CF, Bai LY, Kapuriya N, Peng SY, Wu CY, Sargeant AM, Chen MY, Weng JR. Antitumor effects of BI-D1870 on human oral squamous cell carcinoma. Cancer Chemother. Pharmacol., 73, 237–247 (2014).
- 30) Katayama K, Fujiwara C, Noguchi K, Sugimoto Y. RSK1 protects P-glycoprotein/ABCB1 against ubiquitin-proteasomal degradation by downregulating the ubiquitin-conjugating enzyme E2 R1. Sci. Rep., 6, 36134 (2016).
- 31) Katayama K, Noguchi K, Sugimoto Y. FBXO15 regulates P-glycoprotein/ABCB1 expression through the ubiquitin-proteasome pathway in cancer cells. Cancer Sci., 104, 694–702 (2013).
- 32) Katayama K, Yamaguchi M, Noguchi K, Sugimoto Y. Protein phosphatase complex PP5/PPP2R3C dephosphorylates P-glycoprotein/ABCB1 and downregulates the expression and function. Cancer Lett., 345, 124–131 (2014).
- 33) Takami M, Katayama K, Noguchi K, Sugimoto Y. Protein kinase C alpha-mediated phosphorylation of PIM-1L promotes the survival and proliferation of acute myeloid leukemia cells. Biochem. Biophys. Res. Commun., 503, 1364–1371 (2018).
- 34) ClinicalTrials.gov. “Metformin + cytarabine for the treatment of relapsed/refractory AML.”: ‹https://clinicaltrials.gov/ct2/show/NCT01849276?term=metformin&cond=AML&draw=2&rank=1›, accessed 23 March, 2021.
- 35) Denton D, Kumar S. Autophagy-dependent cell death. Cell Death Differ., 26, 605–616 (2019).
- 36) Schwartz LM. Autophagic cell death during development—ancient and mysterious. Front. Cell Dev. Biol., 9, 656370 (2021).
- 37) Ferrari S, Bandi HR, Hofsteenge J, Bussian BM, Thomas G. Mitogen-activated 70K S6 kinase. Identification of in vitro 40 S ribosomal S6 phosphorylation sites. J. Biol. Chem., 266, 22770–22775 (1991).
- 38) Flotow H, Thomas G. Substrate recognition determinants of the mitogen-activated 70K S6 kinase from rat liver. J. Biol. Chem., 267, 3074–3078 (1992).
- 39) Chae HD, Dutta R, Tiu B, Hoff FW, Accordi B, Serafin V, Youn M, Huang M, Sumarsono N, Davis KL, Lacayo NJ, Pigazzi M, Horton TM, Kornblau SM, Sakamoto KM. RSK inhibitor BI-D1870 inhibits acute myeloid leukemia cell proliferation by targeting mitotic exit. Oncotarget, 11, 2387–2403 (2020).