Abstract
In hepatitis, activated hepatic stellate cells (HSCs) produce collagens, causing liver fibrosis. Microenvironmental stiffness is a known trigger of HSC activation and is communicated through mechanotransduction. Cell proliferation, alpha smooth muscle actin (α-SMA) and collagen type Iα (Col1α) are indicative of activated HSCs. We hypothesized that certain compounds could interfere with the HSC’s recognition of microenvironmental stiffness by blocking cell adhesion signaling. To verify the potential of mechanotransduction, and in particular of focal adhesion proteins, as liver fibrosis drug targets, we evaluated existing drugs. We examined the effects of the integrin antagonist, BS-1417; the focal adhesion kinase (FAK) inhibitor, defactinib; the cyclin-dependent kinase (CDK) inhibitor, roscovitine; and two microtubule modulators, paclitaxel and colchicine, on stiffness-induced HSC activation. To determine the extent of transforming growth factor β (TGF-β) participation in mechanotransduction, we measured gene expression levels of α-SMA and Col1α. We also measured ATP levels to determine cell number. Results revealed that interestingly, although TGF-β did not show additional HSC activation after stiffness stimulation, the TGF-β receptor inhibitor, SB525334, markedly suppressed stiffness-induced α-SMA and Col1α mRNA expression. BS-1417, roscovitine, defactinib and colchicine suppressed α-SMA and Col1α mRNA expression as well as the number of HSCs. Paclitaxel also suppressed stiffness-induced α-SMA mRNA expression and the number of HSCs, but mildly reduced that of Col1α mRNA. Together, these results show that an integrin antagonist and mechanotransduction-targeting drugs reduced stiffness-induced HSC activation in a dose-dependent fashion. The targeting of focal adhesion proteins involved in mechanotransduction is promising in liver fibrosis drug development.
INTRODUCTION
Fibrosis is defined as the excess deposition of extracellular matrix (ECM), mainly collagen, which impairs normal formation and functions in tissues and organs. In the liver, ECM is produced by activated hepatic stellate cells (HSCs), in such a way that HSCs play a key role in the pathogenesis and progression of liver fibrosis. Further, up to 80% of hepatocellular carcinomas develop from liver cirrhosis (fibrosis) following initial pathological injury.1)
Regardless of the cause of the damage, when the liver is injured, quiescent HSCs transdifferentiate into proliferative myofibroblast-like cells which produce ECM. This is termed HSC activation, and defined by various characteristic markers such as upregulation of gene expression of alpha smooth muscle actin (α-SMA) and collagen type Iα (Col1α).2) Cell proliferation is also indicative of activated HSCs. To prevent or reverse liver fibrosis, further elucidation of the HSC activation mechanism and effective drugs are needed. We hypothesized that the compounds evaluated in this study could interfere with the HSC’s recognition of adjacent and nearby cell stiffening that is signaled by cell adhesion via mechanotransduction, which results in quiescent HSCs activating and changing their morphology and properties.
In 2012, the influence of microenvironmental stiffness on cell cultures was already becoming generally accepted.3) In 2016, it was reported that the influence of stiffness in the cells surrounding HSCs, with which the HSC’s have direct or indirect contact, known as the cellular microenvironment, rather than the overall condition of the liver, results in HSC activation through transmembrane receptors called integrin.4,5) Conversely, activated HSCs on stiff plastic dishes can be reversed to a quiescent state, by transplanting them to a soft microenvironment similar to that of normal liver stiffness.6)
Integrins bind to the Arg-Gly-Asp (RGD) motif on the surface of other cells and ECM molecules, where they transmit physical information to HSCs. This transmission of information (mechanotransduction) is mediated through the binding of a number of integrin-binding molecules, such as focal adhesion kinase (FAK).7) It has also been reported that Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) are major factors of mechanotransduction triggered by ECM stiffness8,9) and that YAP/TAZ acts as an intracellular mechanical stiffness sensor.10) We confirmed that in vitro, HSCs reacted differently to normal-liver-stiffness collagen gels (3.2 kPa) and hard polystyrene culture dishes (3 GPa) at Day 8. The α-SMA mRNA level on 3.2 kPa gels was 1.7 arb. units and on polystyrene culture dishes, was 39.9 arb. units. Similarly, the mRNA level of Col1α showed 25.0 arb. units on 3.2 kPa and 68.4 arb. units on 3 GPa.11)
Stiffening of the HSC microenvironment occurs not only in vitro, but also in vivo. Stiffness results from ECM in vivo. In the normal liver, HSCs are quiescent and represent 5–8% of the total number of liver cells.12) They occur intermittently in the space of Disse between the sinusoid and hepatocytes,12) and constantly monitor the physical state of sinusoidal endothelial cells and hepatocytes. Capillarization of the sinusoid, determined by the disappearance of the fenestrae structure and the appearance of basal membranes, which are thin ECM, precedes liver fibrosis.13) This means that the appearance of ECM on the sinusoid occurs under inflammatory conditions and a hardened HSC microenvironment. Further, normally, hepatocytes undergo necrosis following transient injury or normal turnover and are replaced with ECM. New hepatocytes are regenerated in their original site when ECM metabolically decomposes. However, continuous/repeated injury (such as that from diabetes) prevents regeneration of hepatocytes, and increases hepatocyte necrosis. As a result, ECM accumulates in the spaces created by missing hepatocytes and forms hard coatings on the sinusoid, generating microenvironmental stiffness. This stiffness is recognized by quiescent HSCs through mechanotransduction. When HSCs recognize stiffness, they become activated. Activated HSCs proliferate and produce excessive ECM, and other quiescent HSCs react to the accumulated ECM. It is a vicious cycle.14)
In this study, to validate mechanotransduction in stiffness-induced HSC activation as a drug target, and having confirmed it, to reposition drugs related to mechanotransduction in liver fibrosis, we evaluated the efficacy of the integrin antagonist, BS-1417, and 4 cancer drugs: the FAK inhibitor, defactinib; the cyclin-dependent kinase (CDK) inhibitor, roscovitine; and two microtubule modulators, paclitaxel and colchicine; in stiffness-induced HSC activation in vitro. Furthermore, as humoral factors involved in fibrosis, such as transforming growth factor β (TGF-β), have long been anti-fibrosis drug targets, to investigate the involvement of TGF-β in this system, we evaluated TGF-β and the TGF-β1 receptor inhibitor, SB525334, using α-SMA and Col1α mRNA expression levels and ATP levels to determine cell number.
MATERIALS AND METHODS
AnimalsFollowing the animal research guidelines of Kyoto Institute of Technology, we removed livers from and sacrificed normal, 8-week male Sprague-Dawley rats (Slc: SD), obtained from Japan SLC Inc. (Shizuoka, Japan), under 1–3% isoflurane anesthesia, just after their arrival. All animal experiments were approved by the Institutional Animal Care and Use Committee at Kyoto Institute of Technology.
Chemicals and ReagentsThe reagents were obtained from suppliers as follows: rat recombinant TGF-β1 protein (Sino Biological, Beijing, China), SB525334 (Selleck Chemicals, Houston, TX, U.S.A.), roscovitine (Abcam, Cambridge, U.K.), defactinib (Cayman Chemical, Ann Arbor, MI, U.S.A.), paclitaxel and colchicine (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). BS-1417 was synthesized following to the reported procedure.15)
Primary Culture of HSCsNormal rat HSCs were isolated as previously described.6,14) In brief, in situ enzymatic digestion of the liver was performed via sequential perfusion with pronase E (Roche, Basel, Switzerland) and collagenase P (Roche) followed by in vitro digestion in pronase E and collagenase P. The resultant slurry was then diluted and filtered. HSCs were isolated from the total cell population through density gradient centrifugation with Nycodenz (Sigma, St. Louis, MO, U.S.A.) solution.
Primary HSCs were cultured in D-MEM (Nacalai Tesque, Kyoto, Japan) containing 10% fatal calf serum with penicillin and streptomycin on collagen-coated plastic culture dishes. From Day 2 to Day 8, test compounds were added in conditioned medium, and the conditioned medium was changed every 48 h. We observed morphological changes in HSCs microscopically.
Real-Time PCRTotal RNA was isolated from cultured HSCs (RNeasy Mini Kit and ribonuclease (RNase)-Free deoxyribonuclease (DNase) Set, Quiagen, Venlo, the Netherlands). After measurement of tRNA density, the RNA template was reverse transcribed into cDNA (Transcriptor First Strand cDNA Synthesis Kit, Roche), then quantitatively amplified by PCR using PowerUp SYBR Green (Applied Biosystems, Bedford, MA, U.S.A.) in CFX96 Real-Time System (Bio-Rad, Hercules, CA, U.S.A.). PCR was performed with 10 pmol specific primers for rat α-SMA (5′-TGT GCT GGA CTC TGG AGA TG-3′) and (5′-GAA GGA ATA GCC ACG CTC AG-3′), Col1α (5′-ACG TCC TGG TGA AGT TGG TC-3′) and (5′-TCC AGC AAT ACC ATG AGG TC-3′), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (5′-GAC ATG CCG CCT GGA GAA AC-3′) and (5′-AGC CCA G GAT GCC CTT TAG T-3′). Individual real-time PCR reactions were performed in triplicate. Results were analyzed using a CFX Manager. In all measured transcripts, we normalized mRNA expression to rat GAPDH to show the expression level per cell.
Quantitative Determination of ATPWe substituted ATP level for cell number in cell proliferation. The amount of ATP was determined using a Cell-Titer Glo Luminescent Cell Viability Assay (Promega, Madison, WI, U.S.A.).
Statistical AnalysisAll data are expressed as the mean ± standard error (S.E.) of three independent experiments. Differences between the treatment groups were evaluated using one-way ANOVA with a Tukey test. Differences were considered significant at p < 0.05.
RESULTS
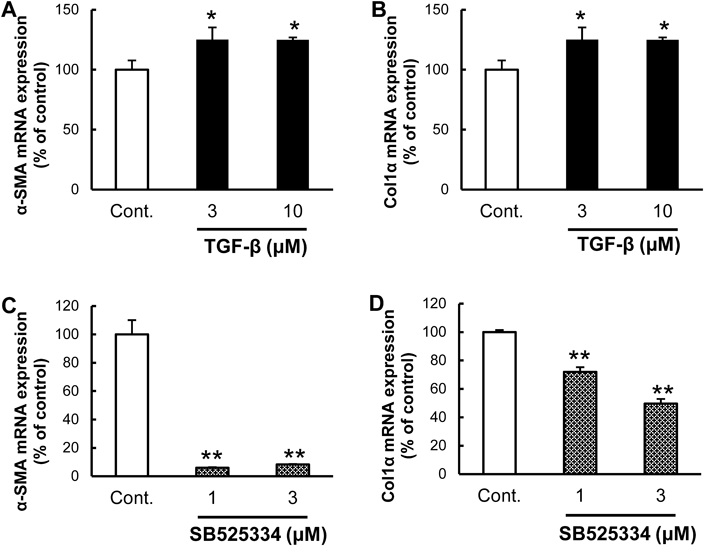
TGF-β Receptor Inhibitor Suppressed Expression of α-SMA and Col1α mRNAAlthough TGF-β caused a significant 25% increase in the mRNA expression level of α-SMA (Fig. 1A) and Col1α (Fig. 1B) at low and high doses, the effects were not dose-dependent. On the other hand, a selective TGF-β receptor inhibitor, SB525334, significantly suppressed the mRNA expression of α-SMA (Fig. 1C) and Col1α (Fig. 1D); respectively. These results suggest that although TGF-β signals are implicated secondarily or in downstream stiffness-induced HSC activation, TGF-β does not promote a significant additive effect on HSCs after stiffness stimulation.
Integrin Antagonist Suppressed Stiffness-Induced HSC ActivationThe efficacy of BS-1417 was evaluated in stiffness-induced HSC activation. BS-1417 reduced the mRNA expression level of α-SMA (Fig. 2A) and Col1α (Fig. 2B) as well as the cell number (Fig. 2C). Micrographs and a representation of the molecular structure of BS-1417 are shown in Figs. 2D and 2E. These results suggest that integrin antagonists reduce stiffness-induced HSC activation.
CDK Inhibitor and FAK Inhibitor Suppressed Stiffness-Induced HSC ActivationThe efficacy of roscovitine and defactinib was evaluated in stiffness-induced HSC activation. Roscovitine and defactinib reduced the mRNA expression level of α-SMA (Fig. 3A) and Col1α (Fig. 3B) as well as cell number (Fig. 3C). These results suggest FAK-related inhibitors reduce stiffness-induced HSC activation.
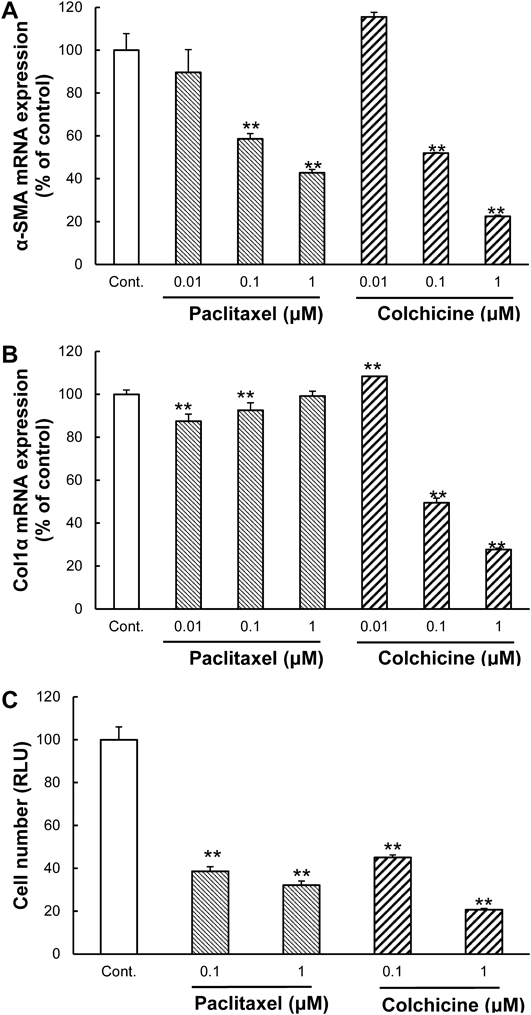
Microtubule Modulators Suppressed Stiffness-Induced HSC ActivationThe efficacy of colchicine and paclitaxel was evaluated following stiffness-induced HSC activation. Both reagents reduced the mRNA expression level of α-SMA (Fig. 4A) and Col1α (Fig. 4B) as well as cell number (Fig. 4C). Although the inhibition rate of paclitaxel in Col1α mRNA expression was 10.4% at a low dose, at higher concentrations, its inhibitory action diminished dose-dependently. These results suggest that microtubule-targeting reagents suppressed stiffness-induced HSC activation. Expression of α-SMA mRNA and cell number showed a similar tendency. Additionally, although increasing α-SMA and Col1α mRNA expression levels are both hallmarks of HSC activation, and could be expected to react in the same way, they did not. It seems that expression of α-SMA mRNA synchronizes with cell number.
DISCUSSION
HSC activation is critical in pathogenesis and progress of liver fibrosis. As mentioned above, a more complete understanding of the precise mechanism of this activation and effective liver fibrosis drugs is required. Because it is known that TGF-β, and the enzymes involved in collagen biosynthesis or degradation, modulate collagen accumulation, they have been considered liver fibrosis drug targets. It is also known that microenvironmental stiffness induces HSC activation. We therefore hypothesized that compounds interfering with any part of the focal adhesions area, could result in an effective fibrosis drug target.
In fact, our data is consistent with the idea that mechanotransduction, the action of stiffness stimuli (in this case) transducing a signal, is a potential therapeutic target in liver fibrosis. The integrin antagonist, FAK inhibitor, CDK inhibitor and microtubule modulators we tested, all suppressed stiffness-induced HSC activation in vitro (Figs. 2–4). The drugs; defactinib, roscovitine, paclitaxel and colchicine, are known to be efficacious in cancer treatment, but our data suggest they are also promising as a therapeutic strategy for liver fibrosis.
In this study we used primary cultured normal HSCs on plastic culture dishes rather than established HSC cell lines which already have proliferative capacity, and the ability to respond to TGF-β, as do transdifferentiated myofibroblast-like cells.16) Unlike primary cultured normal HSCs, established HSC cell lines, such as LX-2, do not respond to stiffness.16) HSC cell lines were first designed or selected in the 1980 s to eliminate the costs and other disadvantages, such as animal dissection, associated with primary culture.17,18) In 2011, it was reported that stiffness caused HSC activation,14) but some years earlier when they were established, TGF-β and enzymes related to collagen biosynthesis or metabolic degradation were some of the only known causes of HSC activation. As response to stiffness was integral to our analysis, we employed primary cultured normal HSCs.
To inspect how TGF-β participates in this system, we evaluated TGF-β and the TGF-β receptor inhibitor. Our results showed that TGF-β does not promote a significant additive effect on HSCs after stiffness stimulation (Figs. 1A, B), and that the TGF-β receptor inhibitor suppressed HSC activation (Figs. 1C, D). The phenomenon of HSC activation is comprised of two events; initially, quiescent HSCs transdifferentiate into myofibroblast-like cells, and then the transdifferentiated cells produce ECM. It is known that TGF-β does not activate quiescent HSCs,6) but acts on fibroblasts,19,20) and that myofibroblasts exhibit autocrine induction of TGF-β.21) Therefore, we suspect TGF-β signals are implicated secondarily or in downstream stiffness-induced HSC activation. If so, TGF-β inhibitors may affect transdifferentiated HSCs by blocking the autocrine action of TGF-β.
Therefore, although the TGF-β receptor inhibitor acts on activated HSCs, this may not extend to inducing activated ones to quiescent state reversion when stiffness stimuli ceases. Conversely, mechanotransduction inhibitors do induce activated HSCs to revert to quiescence, even when stiffness stimuli, a cause of HSC activation, ceases, as explained below.
Using primary cultured normal HSCs on plastic culture dishes, we evaluated inhibitors, concerned with focal adhesions. The integrin antagonist, FAK inhibitor, and the CDK inhibitor suppressed stiffness-induced HSC activation in α-SMA and Col1α mRNA expression as well as cell proliferation (Figs. 2, 3). These results suggest that inhibiting integrin signaling is an efficacious drug target for liver fibrosis.
We suggest that suppressing FAK mediates CDK inhibitor-induced HSC inactivation. In the 1–40 µM range, roscovitin, a pan-CDK inhibitor, suppressed FAK and extracellular signaling-regulated kinase (ERK) 2.22) Integrin regulated FAK-Src signaling, allowing downstream activation of multiple intracellular signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)/Akt, FAK–c-Jun N-terminal kinase (JNK) and mitogen-activated protein kinase (MAPK)/Erk pathways which relate to cell survival, movement, and proliferation in cancer cells.23) In addition, CDK5 combined to form an active complex with FAK and talin.24)
Next, we evaluated two microtubule modulators, paclitaxel and colchicine. Research has showed that paclitaxel inhibits tumor fibrosis25) and pulmonary fibrosis.26) Because microtubules are involved in migration,27) epithelial-to-mesenchymal transition of cancer cells,28) reaction to osmotic pressure and tension29) and focal adhesions when cell movement is initialized30); we hypothesized that microtubules participate in mechanotransduction. Our data showed that microtubule modulators suppressed HSC activation (Fig. 4). These modulators may not have a cytostatic effect on mitotic inhibition, but on modulation of mechanotransduction-related microtubules. Additionally, mechanotransduction inhibitors, unlike the TGF-β receptor inhibitor, may cause activated HSCs to revert to a quiescent state even when stiffness stimulus ceases.
Although both α-SMA and Col1α are myofibroblastic markers, paclitaxel had an opposite effect on their respective mRNA expression levels (Figs. 4A, B). This tendency was also observed in SB525334 (Figs. 1C, D) and defactinib (Figs. 3A, B). As mRNA expression of α-SMA, rather than that of Col1α, seems to synchronize with cell number, mRNA expression levels of α-SMA and Col1α can be said to reflect morphology and function of HSCs, respectively. It follows that because altered gene expression of an ECM, such as collagen, correlates with the severity of the pathological state in early-stage liver fibrosis, it may function as a biomarker of the fibrotic state. The most accurate marker for the fibrotic state, therefore, is not altered gene expression of α-SMA, but collagen.
In conclusion, our data showed integrin signaling inhibition and microtubule modulation suppressed stiffness-induced HSC activation in a dose-dependent manner, and suggest a promising anti-fibrosis drug target: mechanotransduction. Cancer drugs involved in mechanotransduction can be repositioned as antifibrotic drugs.
Acknowledgment
We thank Ms. Wanda Miyata for help with critical review.
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
The online version of this article contains supplementary materials.
REFERENCES
- 1) Hernandez-Gea V, Toffanin S, Friedman SL, Llovet JM. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology, 144, 512–527 (2013).
- 2) De Minicis S, Seki E, Uchinami H, Kluwe J, Zhang Y, Brenner DA, Schwabe RF. Gene expression profiles during hepatic stellate cell activation in culture and in vivo. Gastroenterology, 132, 1937–1946 (2007).
- 3) Kolahi KS, Donjacour A, Liu X, Lin W, Simbulan RK, Bloise E, Maltepe E, Rinaudo P. Effect of substrate stiffness on early mouse embryo development. PLOS ONE, 7, e41717 (2012).
- 4) Desai SS, Tung JC, Zhou VX, Grenert JP, Malato Y, Rezvani M, Español-Suñer R, Willenbring H, Weaver VM, Chang TT. Physiological ranges of matrix rigidity modulate primary mouse hepatocyte function in part through hepatocyte nuclear factor 4 alpha. Hepatology, 64, 261–275 (2016).
- 5) Zhubanchaliyev A, Temirbekuly A, Kongrtay K, Wanshura LC, Kunz J. Targeting machanotransduction at the transcriptional level: YAP and BRD4 are novel therapeutic targets for the reversal of liver fiblosis. Front. Pharmacol., 7, 462 (2016).
- 6) Caliari SR, Perepelyuk M, Soulas EM, Lee GY, Wells RG, Burdick JA. Gradually softening hydrogels for modeling hepatic stellate cell behavior during fibrosis regression. Integr. Biol. (Camb.), 8, 720–728 (2016).
- 7) Tsimbouri PM. Adult stem cell responses to nanostimuli. J. Funct. Biomater., 6, 598–622 (2015).
- 8) Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, Zanconato F, Le Digabel J, Forcato M, Bicciato S, Elvassore N, Piccolo S. Role of YAP/TAZ in mechanotransduction. Nature, 474, 179–183 (2011).
- 9) Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, Dupont S, Piccolo S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell, 154, 1047–1059 (2013).
- 10) Yang C, Tibbitt MW, Basta L, Anseth KS. Mechanical memory and dosing influence stem cell fate. Nat. Mater., 13, 645–652 (2014).
- 11) Sakai M, Sumiyoshi T, Aoyama T, Urayama K, Yoshimura R. GPR91 antagonist and TGF-β inhibitor suppressed collagen production of high glucose and succinate induced HSC activation. Biochem. Biophys. Res. Commun., 530, 362–366 (2020).
- 12) Geerts A. History, heterogeneity, developmental biology, and function of quiescent hepatic stellate cells. Semin. Liver Dis., 21, 311–336 (2001).
- 13) DeLeve LD, Wang X, Guo Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology, 48, 920–930 (2008).
- 14) Olsen AL, Bloomer SA, Chan EP, Gaça MDA, Georges PC, Sackey B, Uemura M, Janmey PA, Wells RG. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am. J. Physiol. Gastrointest. Liver Physiol., 301, G110–G118 (2011).
- 15) Iwama S, Kitano M, Fukuya F, Honda Y, Sato Y, Notake M, Morie T. Discovery of a potent and selective αvβ3 integrin antagonist neointima formation in rat balloon injury model. Bioorg. Med. Chem. Lett., 14, 2567–2570 (2004).
- 16) Xu L, Hui AY, Albanis E, Arthur MJ, O’Byrne SM, Blaner WS, Mukherjee P, Friedman SL, Eng FJ. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut, 54, 142–151 (2005).
- 17) Borojevic R, Monteiro AN, Vinhas SA, Domont GB, Mourão PAS, Emonard H, Grimaldi G Jr, Grimaud JA. Establishment of a continuous cell line from fibrotic schistosomal granulomas in mice livers. In Vitro Cell. Dev. Biol., 21, 382–390 (1985).
- 18) Herrmann J, Gressner AM, Weiskirchen R. Immortal hepatic stellate cell lines: useful tools to study hepatic stellate cell biology and function? J. Cell. Mol. Med., 11, 704–722 (2007).
- 19) Ghahary A, Shen YS, Scott PG, Tredget EE. Immunolocalization of TGF-β1 in human hypertrophic scar and normal dermal tissues. Cytokine, 7, 184–190 (1995).
- 20) Tredget EE, Wang R, Shen Q, Scott PG, Ghahary A. Transforming growth factor-beta mRNA and protein in hypertrophic scar tissues and fibroblasts: antagonism by IFN-alpha and IFN-gamma in vitro and in vivo. J. Interferon Cytokine Res., 20, 143–152 (2000).
- 21) Dabiri G, Campaner A, Morgan JR, Van De Water L. A TGF-β1-dependent autocrine loop regulates the structure of focal adhesion in hypertrophic scar fibroblasts. J. Invest. Dermatol., 126, 963–970 (2006).
- 22) Bach S, Knockaert M, Reinhardt J, et al. Roscovitine targets, protein kinases and pyridoxal kinase. J. Biol. Chem., 280, 31208–31219 (2005).
- 23) Aksorn N, Chanvorachote P. Integrin as a molecular target for anti-cancer approaches in lung cancer. Anticancer Res., 39, 541–548 (2019).
- 24) Liang Q, Li L, Zhang J, Lei Y, Wang L, Liu DX, Feng J, Hou P, Yao R, Zhang Y, Huang B, Lu J. CDK is essential for TGF-β1-induced epithelial–mesenchymal transition and breast cancer progression. Sci. Rep., 3, 2932 (2013).
- 25) Tsukada T, Fushida S, Harada S, Terai S, Yagi Y, Kinoshita J, Oyama K, Tajima H, Ninomiya I, Fujimura T, Ohta T. Low-dose paclitaxel modulates tumor fibrosis in gastric cancer. Int. J. Oncol., 42, 1167–1174 (2013).
- 26) Wang C, Song X, Li Y, Han F, Gao S, Wang X, Xie S, Lv C. Low-dose paclitaxel ameliorates pulmonary fibrosis by suppressing TGF-β1/Smad3 pathway via miR-140 upregulation. PLOS ONE, 8, e70725 (2013).
- 27) Ganguly A, Yang H, Sharma R, Patel KD, Cabral F. The role of microtubules and their dynamics in cell migration. J. Biol. Chem., 287, 43359–43369 (2012).
- 28) Whipple RA, Matrone MA, Cho EH, Balzer EM, Vitolo MI, Yoon JR, Ioffe OB, Tuttle KC, Yang J, Martin SS. Epithelial-to-mesenchymal transition promotes tubulin detyrosination and microtentacles that enhance endothelial engagement. Cancer Res., 70, 8127–8137 (2010).
- 29) Kaverina I, Krylyshkina O, Beningo K, Anderson K, Wang YL, Small JV. Tensile stress stimulates microtubule outgrowth in living cells. J. Cell Sci., 115, 2283–2291 (2002).
- 30) Matsumoto S, Fumoto K, Okamoto T, Kaibuchi K, Kikuchi A. Binding of APC and dishevelled mediates Wnt5a-regulated focal adhesion dynamics in migrating cells. EMBO J., 29, 1192–1204 (2010).