Abstract
Delavatine A (DA) is an unusual isoquinoline alkaloid with a novel skeleton isolated from Chinese folk medicine Incarvillea delavayi. Studies conducted in our lab have demonstrated that DA has potential anti-inflammatory activity in lipopolysaccharide (LPS)-treated BV-2 cells. DA, however, has not been studied for its protective effect on neuronal cells yet. Thus, to explore whether DA can protect neurons, oxygen and glucose deprivation/reperfusion (OGD/R)-injured PC12 cell and middle cerebral artery occlusion/reperfusion (MCAO/R) rat model were used to assess the protective efficacy of DA against OGD/R damaged PC12 cells and MCAO/R injured rats. Our results demonstrated that DA pretreatment (0.31–2.5 µM) dose-dependently increased cell survival and mitochondrial membrane potential (MMP), whereas it lowered the leakage of lactate dehydrogenase (LDH), intracellular cumulation of Ca2+, and overproduction of reactive oxygen species (ROS), and inhibited the apoptosis rate in OGD/R-injured PC12 cells. Western blot demonstrated that DA pretreatment lowered the expression of apoptotic proteins and repressed the activation of the mitogen-activated protein kinase kinase 7 (MKK7)/c-Jun N-terminal kinase (JNK) pathway. It was also found that the neuroprotective efficacy of DA was significantly reversed by co-treatment with the JNK agonist anisomycin, suggesting that DA reduced PC12 cell injury and apoptosis by suppressing the MKK7/JNK pathway. Furthermore, DA oral administration greatly alleviated the neurological dysfunction and reduced the infarct volume of MCAO/R rats. Taken together, DA could ameliorate OGD/R-caused PC12 cell injury and improve brain ischemia/reperfusion (I/R) damage in MCAO/R rats, and its neuroprotection might be attributed to suppressing the MKK7/JNK signaling pathway.
INTRODUCTION
Stroke is the world’s main cause of physical disability, which brings about a tremendous global economic burden annually.1) Ischemic strokes, which are caused by embolic or thrombotic occlusions in the cerebral vessels, account for about 80% of stroke patients.2) Currently, the only drug approved by the U.S. Food and Drug Administration (FDA) to treat ischemic stroke is intravenous tissue plasminogen activator (tPA).3) Nevertheless, tPA therapy must be administered within 4.5 h following a stroke, and merely a small percentage of stroke patients receive tPA on account of its narrow therapeutic time window.4) Although recannalization therapy based on pharmacological and clinical thrombolysis has been widely applied, neuroprotective agents are beneficial for preventing brain damage following stroke by prolonging the therapeutic time window and ameliorating neural function.5) Therefore, the search for new neuroprotective agents remains urgent.
Oxygen and glucose deprivation (OGD) is one pathophysiological consequence of ischemic stroke, causing excitotoxicity, Ca2+ overload, oxidative stress, and inflammation, ultimately causing neural cell death.6) Cerebral ischemia/reperfusion (I/R) insult usually triggers neural cell apoptosis and necroptosis.7) The c-Jun N-terminal kinase (JNK), which is one of the signaling hubs in the mitogen-activated protein kinase (MAPK) pathway, partakes in numerous biological processes, including cell division, proliferation, and apoptosis.8) Accumulating studies have demonstrated that phosphorylated JNKs may be associated with neuronal death and apoptosis after stroke insult.9–11) In animal experiments, inhibition of JNK can reduce infarct volume and improve neurologic outcomes.12–14) Mitogen-activated protein kinase kinase 7 (MKK7) acts as an upstream activator of JNK, being in charge of JNK’s pathological activation when a cell is stressed.15,16) Targeting MKK7 offers a potential approach to treat neurological illnesses caused by the activation of MKK7/JNK.17) Therefore, suppressing the MKK7/JNK signaling pathway may be a practicable way to ameliorate I/R damage in the brain.
Plant-derived natural products have multiple biological efficacies, such as antioxidative, anti-apoptotic, and anti-inflammatory properties, supporting their prospects for the treatment of stroke.18) Numerous neuroprotective agents have shown protective functions in animal experiments by reducing secondary injury to penumbra tissue or minimizing damage before and after reperfusion to promote neural recovery and plasticity.19,20) Delavatine A (DA, the chemical structure as shown in Fig. 1A) is an unusual isoquinoline alkaloid with a novel carbon skeleton, was isolated from medicinal plant Incarvillea delavayi and has been totally synthesized in our lab.21) Our foregone investigation has revealed that DA alleviated lipopolysaccharide (LPS)-induced BV-2 cell inflammation by inhibiting the nuclear factor-kappa B (NF-κB) pathway to reduce pro-inflammatory cytokine release.22) However, the neuroprotective efficacy of DA remains untested for the treatment of cerebral I/R insult.
Therefore, we aim to explore the protective efficacy of DA on OGD/R-damaged PC12 cells (a cell line with neuronal-like properties) and middle cerebral artery occlusion/reperfusion (MCAO/R) rats, as well as whether DA can suppress PC12 cell apoptosis caused by OGD/R through inhibiting the MKK7/JNK pathway.
MATERIALS AND METHODS
ReagentsDelavatine A (purity > 98%) was synthesized and identified in our lab. For experiments, 10 mM DA was freshly dissolved in dimethyl sulfoxide (DMSO) and diluted with culture medium. Meanwhile, CCK-8, lactate dehydrogenase (LDH), reactive oxygen species (ROS), Fluo-3 AM, JC-1 and BCA were obtained through Beyotime Biotechnology (Shanghai, China). Primary antibodies against β-actin mouse (#3700), MKK7 rabbit (#4172), p-MKK7 rabbit (Ser271/Thr275) (#4171), caspase-3 (D3R6Y) rabbit (#14220), and cleaved caspase-3 (Asp175) rabbit (#9661) were bought from Cell Signaling Technology (Denver, MA, U.S.A.). Primary antibodies against anti-JNK rabbit (ab179461), anti-phospho-JNK rabbit (ab76572), anti-Bcl-2 rabbit (ab182858), and anti-Bax rabbit (ab182733), were obtained from Abcam (Cambridge, U.K.). The IRDye 800CW donkey anti-rabbit (926-32211) and donkey anti-mouse (926-32211) secondary antibodies were acquired from LI-COR (Lincoln, NE, U.S.A.). The JNK agonist anisomycin was obtained from Beyotime Biotechnology.
Cell Cultures and OGD/R-Injured Cell ModelHighly differentiated PC12 cell line was acquired from the Stem Cell Bank (Shanghai, China) and maintained in complete Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, TX, U.S.A) comprising 10% fetal bovine serum in a humidified incubator with 5% carbon dioxide at 37 °C.
In order to imitate in vitro ischemic/reperfusion-like conditions, PC12 cells were submitted to OGD/R induction. Concisely, cells were washed twice with phosphate buffered saline (PBS) gently after being cultured and adhered for 24 h, and maintained in a deoxygenated glucose-free DMEM (Invitrogen) under an anaerobic incubator (1% oxygen, 94% nitrogen, 5% carbon dioxide) at 37 °C for 90 min. Afterwards, the cells were transferred to a normal atmosphere and maintained in complete DMEM for 24 h. Untreated cells were cultured in complete DMEM under routine conditions.
Cell MorphologyThe morphological changes of PC12 cells were assessed after OGD/R treatment with or without indicated concentrations of DA and Ani. The cell body volume was quantified by cell area per microscope field with Image J software.
CCK-8 Screening Determined the Viability of CellsConcisely, the cells (1 × 104 /well) were cultured and adhered in a 96-well plate for 24 h. After pretreatment with the selected concentrations of DA for 2 h, the cells were washed twice with PBS gently, submitted to OGD for 90 min, and reoxygenated for 24 h. Afterward, cells were cultured for 30 min at 37 °C following the addition of 10 mL of CCK-8 solution. The absorbance measurement was carried out using a Bio-Tek microplate reader at 450 nm (Winooski, VT, U.S.A.).
LDH Release Assessed Cell CytotoxicityBriefly, following OGD/R exposure, the amount of LDH was measured based on the product instructions. The absorbance of 490 nm was examined using a Bio-Tek microplate reader. The amount of LDH in the culture medium was presented as a percent of total LDH.
Examination of ROS LevelsBriefly, after OGD/R treatment, PC12 cells were cultured with 10 mM 2′,7′-dichlorofluorescein diacetate (DCFH-DA) at 37 °C for 30 min, and twice washed with PBS. Fluorescence pictures were taken employing a fluorescence microscope and the intensity of fluorescence for ROS was recorded at an emission of 525 nm wavelength upon excitation at 488 nm using a POLARstar Omega plate reader (BMG Labtech).
Measurement of MMPMMP was detected by JC-1 dye staining. Concisely, following OGD/R exposure, PC12 cells were co-incubated with JC-1 for 20 min at 37 °C following the manufacturer's instructions. Fluorescence pictures were taken employing a fluorescence microscope. The ratio of red-to-green fluorescence indicated an early phase of cell apoptosis after OGD/R injury.
Inspection of Intracellular Ca2+ LevelsIntracellular accumulation of Ca2+ was determined using the Fluo-3 AM assay. Briefly, following OGD/R damage, PC12 cells were cultured with 5 mM Fluo-3 AM at 37 °C for 60 min following product instructions. Fluo-3 AM fluorescence was determined using a POLARstar Omega plate reader.
Flow Cytometry ScreeningThe PC12 cell apoptosis was detected by the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) detection kit (Dojindo, Kumamoto, Japan). Concisely, PC12 cells (4 × 105 cells per well) were adhered to 6-well plates for 24 h. Following OGD/R exposure, cells were harvested and suspended in 1 × binding buffer, and incubated with Annexin V-FITC/PI at 25 °C for 20 min. Apoptotic cell rates were described as the percentages of early apoptotic cells (Annexin V+/PI−) and late apoptotic cells (Annexin V+/PI+) using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, U.S.A.).
Western Blotting ExperimentsCells were lysed using NP-40 (Beyotime) buffer comprising 1 × Halt™ protease and phosphatase inhibitor cocktail (Thermo Scientific), and quantified by BCA (Beyotime). Equivalent amounts of protein (20 µg) were blended with loading buffer, heated to boiling for 5 min, separated using a 10.0–12.5% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) gel, and transferred to a polyvinylidene difluoride (PVDF) membrane. The membranes were blocked with 1 × protein-free blocking buffer (Sangon Biotech, China) for 5.0 min at 25 °C, and then incubated overnight at 4 °C with diluted primary antibodies including p-JNK (1 : 1000), JNK (1 : 1000), p-MKK7 (1 : 1000), MKK7 (1 : 1000), caspase-3 (1 : 1000), cleaved caspase-3 (1 : 1000), Bax (1 : 1000), Bcl-2 (1 : 1000), and β-actin (1 : 10000). The membranes were washed and incubated with diluted IR dye-labeled secondary antibodies for 1 h at room temperature. The membranes were detected and the blots were analyzed by employing the Odyssey IR imaging system.
Animals and Experimental GroupsAnimal experiments were performed following the Guide for Care and Use of Laboratory Animals of Naval Medical University and approved by the Committee on Ethics of Medicine, Naval Medical University. Male Sprague Dawley (SD) rats (250–270 g) were bought from BiKai Biotechnology Co., Ltd. (Shanghai, China) and maintained in a certified animal facility. The experimental animals were separated into four groups (n = 6 each) in a random manner: (1) the sham group; (2) the MCAO/R + vehicle group; (3) the MCAO/R + 5 mg/kg DA; (4) the MCAO/R + 10 mg/kg DA. DA was dissolved in CMC-Na and diluted to the indicated concentrations. The rats were orally administered with DA or CMC-Na for four consecutive days, followed by the establishment of the MCAO/R model on the fourth day.
The Construction of the MCAO/R Model in VivoThe MCAO/R model was constructed as previously reported.23) Concisely, the rats were intraperitoneally injected with sodium pentobarbital (40 mg/kg) for deep anesthesia throughout the operation. The right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were exposed by a blunt dissection procedure. The head diameter of 0.36 ± 0.02 nylon suture (Xinong, China) was inserted into the CCA to obstruct the middle cerebral artery for 2 h, and then the suture was taken out to reperfusion for 24 h. The sham group received the same surgery but without occlusion.
Neurological Deficit Scoring and Measurement of Infarct VolumesNeurological deficits were assessed 24 h after MCAO/R by using the Zea–Longa scoring system.
Table 1 The Scoring Standard of the Neurological Deficit Evaluation
| Score | Scoring standard |
|---|
| 0 | No apparent neurological dysfunction |
| 1 | Unable to extend the affected forelimb |
| 2 | Walking in circles |
| 3 | Tumbling to the side while walking because of hemiplegia |
| 4 | Unconscious and unable to walk |
The rats were anesthetized and sacrificed after behavioral evaluation. Brain slices were cut into five 2 mm thick coronal slices after the brain was removed. Slices were stained for 20 min at 37 °C with 1.0% TTC in a 6-well plate in the dark, followed by immersion in 4% formaldehyde overnight. The infarct area was calculated for each slice employing Image J software. The infarct volume was calculated as follows:
Statistical AnalysisData was analyzed using GraphPad Prism 7.0 software and displayed as mean ± standard deviation (S.D.). To determine statistically significant differences between multiple groups, a one-way ANOVA followed by Tukey’s multiple comparisons test was applied. The difference of p < 0.05 means statistical significance.
RESULTS
OGD/R Time-Dependently Inhibited PC12 Cell SurvivalTo select the OGD exposure time, PC12 cells were exposed to an OGD environment for the indicated time periods (0, 30, 60, 90, 120 min) and then reoxygenated for 24 h. As demonstrated in Fig. 1B, the viability of PC12 cells was time-dependently decreased. The viability was reduced to about 50% after OGD exposure for 90 min. Thus, 90 min of OGD-induction time was selected for subsequent experiments.
DA Pretreatment Raised PC12 Cell Survival and Prevented LDH ReleaseThe cytotoxicity and protecting effect of DA were analyzed with a CCK-8 assay. As illustrated in Fig. 1C, DA did not show obvious cytotoxicity against PC12 cells with an IC50 value of 85.32 ± 0.12 µM at 24 h. The concentration range of 0–6.25 µM DA did not affect PC12 cell viability. Thus, DA at concentrations of 0.3–5.0 µM were used for subsequent studies.
Figure 1D illustrated that the cell viability of the OGD/R group was remarkably decreased. Pretreatment with DA (0.31–5.0 µM) notably increased PC12 cell viability, in which 5.0 µM DA showed no better protective effect than that of 2.5 µM DA. This result was confirmed by the LDH release screening. As displayed in Fig. 1E, OGD/R exposure notably increased LDH release, whereas DA pretreatment (0.31–5.0 µM) significantly attenuated this effect. Pretreatment with 5.0 µM DA did not show stronger protection than 2.5 µM DA in the LDH release assay. Based on these results, we chose 2.5 µM DA as the highest dose for subsequent experiments. Accordingly, the concentration of the DA-alone group was adjusted to be 2.5 µM, too.
In addition, cell morphology analysis demonstrated that PC12 cells in the control group appeared long and striped, had long axons, and were arranged in regular pattern. When being exposed to OGD/R, the injured cells were shrunken and disarranged, and even the cell number appeared to decrease. Pretreatment with DA (0.5, 1.0, and 2.5 µM) significantly increased cell volume (Fig. 1F), which manifested in neuronal synapses lengthening, soma enlarging (Fig. 1G). DA-only treatment groups alone did not affect PC12 cell morphology.
DA Pretreatment Decreased the Intracellular Amount of Ca2+, Attenuated ROS Levels, Reversed the Potential Level of the MMP, and Repressed Apoptosis in PC12 Cells Undergone OGD/RDuring ischemia, excessive Ca2+ influx into cells triggers ROS overproduction and mitochondrial dysfunction, leading to neuronal apoptosis and necrosis.24) We measured the accumulation of intracellular Ca2+ in OGD/R-damaged PC12 cells using a fluorescence spectrophotometer. As illustrated in Fig. 2B, OGD/R exposure remarkably increased the accumulation of Ca2+ in PC12 cells. Pretreatment with DA (0.31–2.5 µM) dose-dependently reduced intracellular Ca2+ levels. Treatment with 2.5 µM DA alone did not affect the level of Ca2+ in normal PC12 cells.
Similarly, the intracellular ROS levels were measured using fluorescence microscopy and a spectrophotometer, respectively. The OGD/R group displayed a strong green fluorescence intensity of ROS as illustrated in Fig. 2A (I). However, DA treatment (0.5, 1.0, and 2.5 µM) obviously decreased the fluorescence intensity. The result was verified by a fluorescence spectrophotometer (Fig. 2C). The OGD/R exposure gave rise to the elevation of the ROS level. Pretreatment with DA (0.31–2.5 µM) dose-dependently decreased the ROS generation in OGD/R-damaged PC12 cells, whereas DA-only treatment did not influence the ROS level in the normal group. Results showed that DA could evidently decrease intracellular Ca2+ and ROS levels in PC12 cells after OGD/R.
Ischemic stroke is closely related to mitochondrial dysfunction, which may result in mitochondrial permeability transition pore (mPTP) opening, MMP collapse, and cytochrome c release, all of which induce cell death.25) JC-1 was employed to inspect the change of MMP, which indirectly reflects the degree of mPTP opening. As shown in Figs. 2A (II) and D, the MMP level after OGD/R exposure was significantly reduced. In contrast, pretreatment with DA (0.5, 1.0, and 2.5 µM) significantly elevated the MMP level, whereas treatment with DA alone did not affect the MMP level. This result indicated that DA could enhance the MMP levels of OGD/R-induced PC12 cells.
Additionally, apoptosis was examined based on flow cytometry. As seen in Figs. 2E and F, 2.5 µM DA did not change the apoptosis rates of the undamaged PC12 cells. After undergoing exposure to OGD/R, the apoptosis rate was increased to 13.50%. However, DA pretreatment (1.0 and 2.5 µM) significantly reduced the apoptosis rates of the OGD/R-injured PC12 cells to 7.14 and 5.51%, respectively, whereas no change was observed for the 0.5 µM DA treated group. This result showed that DA could reduce PC12 cell apoptosis percentage after OGD/R.
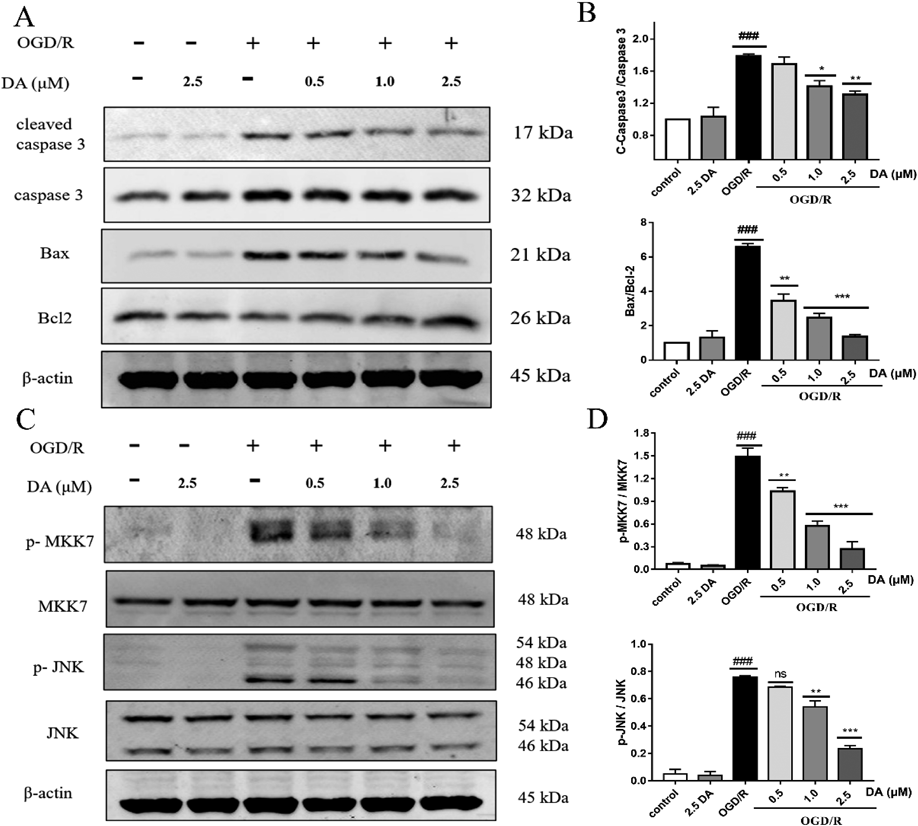
Pretreatment with DA Lowered the Levels of Apoptosis Proteins and Suppressed the Activation of the MKK7/JNK Signal Pathway in PC12 Cells Following OGD/RBy Western blotting, we examined the expression of the apoptosis-related protein. OGD/R exposure significantly elevated the levels of the cleaved caspase 3 and Bax, whereas it decreased Bcl-2 protein expression, resulting in the higher Bax/Bcl-2 ratio. Pretreatment with DA dose-dependently reduced the expression of cleaved caspase 3 and Bax, and increased Bcl-2 expression (Figs. 3A, B). This result demonstrated that DA could reduce the expression of apoptotic proteins in PC12 cells exposed to OGD/R.
JNKs belong to the MAPK family and can be activated by MKK4 and MKK7. MKK7 has been proven to be a specific activator of JNK. Thus, under OGD/R injury, the protein expression of MKK7 and JNK were examined. The OGD/R group exhibited increased p-MKK7 and p-JNK, while pretreatment of DA dose-dependently lowered the levels of p-MKK7 and p-JNK (Figs. 3C, D). This result implied that DA could significantly inhibit the activation of the MKK7/JNK pathway in PC12 cells that have undergone OGD/R damage.
DA Pretreatment Lessened Cell Damage and Apoptosis via Inhibiting the MKK7/JNK Pathway in PC12 Cells Undergoing OGD/RTo investigate whether suppression of the JNK pathway mediates the neuroprotection of DA, 5.0 µM anisomycin (Ani, a specific agonist of the JNK pathway) was employed in this study.9) As seen in Figs. 4 and 5, after Ani treated the exposed and unexposed OGD/R, the cell viability was decreased, the apoptosis rate was increased, the levels of proapoptotic proteins were elevated, and the MKK7/JNK pathway was activated, suggesting Ani could activate the MKK7/JNK pathway to increase cell injury and apoptosis in PC12 cells. After co-administration of DA and Ani, the effect of DA against OGD/R-caused cell morphology changes and injury was markedly abolished by the JNK agonist Ani relative to the OGD/R + DA group (Figs. 4A–C). Similarly, compared with those of the OGD/R + DA group, co-treatment with DA and Ani not only increased cell apoptosis rate (Figs. 4D, E), but also raised the amount of p-MKK7, p-JNK, and cleaved caspase 3 as well as Bax/Bcl-2 ratio in PC12 cells after OGD/R (Fig. 5), suggesting that Ani eliminated the anti-apoptotic effects of DA against OGD/R-damaged PC12 cells. These results showed that the protective efficacy of DA was markedly reversed by the JNK agonist Ani, indicating that DA might attenuate the OGD/R-induced cell damage and apoptosis by inhibiting the MKK7/JNK pathway.
DA Administration Ameliorated the Neurological Deficit and the Reduced Infarct Volume of MCAO/R RatsFurther evaluation of the neuroprotective effect of DA was conducted using the MCAO/R model in vivo. None of the animals died during this operation. Figure 6A illustrates the experimentation scheme. After 24 h of reperfusion, the MCAO/R group demonstrated severe neurological deficits. DA administration at the dose of 10 mg/kg remarkably lowered the neurological deficit score (Fig. 6B).
The volume of cerebral infarction was assessed by TTC staining. The MCAO/R group showed obvious infarction. In contrast, administration with DA (5 and 10 mg/kg) markedly lessened the infarct volume of MCAO/R rats (Figs. 6C, D), suggesting that DA appears to be effective in protecting rats from MCAO/R.
DISCUSSION
Stroke is a serious cerebrovascular disease associated with large financial and social costs for families and communities. In the treatment of stroke, reperfusion and neuroprotection are two main therapeutic strategies. Due to the narrow treatment window, thrombolysis and thrombectomy are commonly limited to a few patients, and these treatments fail to protect patients from ischemic injury.26) Neuroprotective agents have the potential to extend the therapeutic time window of thrombolytic and thrombectomy therapies while also improving neuron survival in the ischemic area and restoring post-ischemic stroke neural function.5) So, it is still very important for stroke treatment to discover novel neuroprotective agents.
Natural products are a valuable source of drug discovery, with potential benefits in stroke treatment by interfering with excitotoxicity, oxidative and nitrifying stress, inflammation, and other pathological processes.18) Delavatine A, a newly discovered isoquinoline alkaloid, has been isolated from the folk medicinal plant Incarvillea delavayi. Previous studies conducted in our group have revealed that DA could suppress the NF-κB pathway in LPS-stimulated BV-2 cells and show an effective anti-inflammatory effect.22) Here, the OGD/R injured PC12 cells and MCAO/R damaged rat models were used to further investigate the neuroprotective effect of DA against cerebral I/R damage.
Cerebral artery occlusion causes severe OGD and triggers a string of pathological events that result in irretrievable cerebral injury.24) The OGD/R injured PC12 cells have been frequently employed to construct in vitro ischemia/reperfusion models for assessing the neuroprotective effects of drugs.27,28) The current study successfully established an OGD/R-injured cell model by exposing PC12 cells to OGD for 90 min. In previous studies, OGD/R has been shown to decrease cell survival and increase the release of LDH in cells.29) The current study exhibited that DA pretreatment could effectively increase cell survival and prevent LDH release, suggesting that DA possesses a potent protective efficacy against PC12 cells undergoing OGD/R damage.
The post-ischemic OGD brings about depletion of ATP, impairment of the Na+/K+ ATPase pump, reduction of neuronal membrane potential, and the release of additional glutamate. Over-activated glutamate receptors result in Ca2+ influx, excessive ROS levels, mitochondrial dysfunction, and eventually cell death.24,30) Studies have indicated that Ca2+ overload triggers mitochondrial damage and causes mitochondrial swell and the release of apoptogenic factors.31) Oxidative stress occurs when ROS production is imbalanced with antioxidant defense mechanisms, which in turn influences the pathological process of an ischemic stroke.32) In addition, oxidative stress and mitochondrial Ca2+ overload cause mPTP to open and lead to excessive ROS generation. The association between Ca2+ accumulation and ROS production, as well as mitochondrial damage, indicates that ROS and Ca2+ are important players in ischemic stroke.30,32,33) Our study showed that DA pretreatment notably alleviated the intracellular accumulation of Ca2+ and ROS, suggesting that DA can maintain intracellular Ca2+ homeostasis and reduce ROS overproduction in OGD/R-damaged PC12 cells.
Mitochondrial dysfunction is characterized by loss of MMP and opening of the mPTP, which initiates mitochondria-related apoptosis. The decreased MMP is thus considered to be an early indicator of cell apoptosis.34) Increasing evidence suggests that the B-cell lymphoma family has a critical influence on neuronal death and the apoptotic cascade in cerebral ischemic stroke. When Bax and Bak, two important pro-apoptotic proteins in the Bcl-2 family, are translocated into mitochondria, it reduces MMP and releases cytochrome c, which activates caspase 9 and caspase 3 and initiates signal pathways for cell death.6) Studies have shown that OGD/R injury causes the collapse of MMP and thereby activates the mitochondria-mediated apoptosis pathway.32,35) Our study showed that DA treatment dramatically increased MMP, decreased cell apoptosis percentage, inhibited the expression of cleaved caspase 3 and Bax proteins, and up-regulated Bcl-2 level. These results indicate that DA protects OGD/R injured PC12 cells by inhibiting cell apoptosis.
Accumulating data demonstrates that the JNK pathway gets involved in cerebral I/R insult.10) MKK7 and MKK4 are direct upstream activators of JNK. MKK7 activates JNK exclusively, whereas MKK4 activates p38 MAPK and JNK.36) Both in vitro and in vivo, MKK7 primarily takes responsibility for JNK overactivation during neural excitotoxicity, while MKK4 is mostly in charge of JNK’s physiological role.37) JNK overactivation causes inflammation, oxidative stress, and apoptosis response, so JNK inhibitors help to improve I/R injury.8) Targeting MKK7 represents a new therapeutic strategy against cerebral ischemic stroke and other diseases involving MKK7/JNK activation.17) Several studies have shown that the regulation of the MKK7/JNK pathway ameliorates I/R injury by reducing cell apoptosis.38,39) Our results demonstrated that DA lowered the amount of phosphorylated MKK7 and JNK, thereby inhibiting the activation of the MKK7/JNK pathway in OGD/R-induced PC12 cells. This data demonstrates that the neuroprotective efficacy of DA may be mediated by the inhibition of MKK7/JNK signaling. To confirm the above deduction, a JNK signaling pathway agonist (anisomycin) in combination with DA was employed to co-treat PC12 cells undergoing OGD/R. Results showed that anisomycin eliminated the benefits of DA, as characterized by PC12 cell morphological changes, a weakened viability, an elevated apoptosis percentage, and activation of the MKK7/JNK pathway after OGD/R. These results demonstrate that DA might reduce the injury and apoptosis of PC12 cells exposed to OGD/R by suppressing the MKK7/JNK pathway.
To further evaluate the neuroprotective potential of DA, a transient MCAO/R rat model was utilized to mimic human ischemic stroke in vivo.23,40,41) Previous research has reported that the MCAO/R rats will exhibit an elevated neurological deficit score and enlarged infarct volume similar to those of stroke patients.13,17,20) Neurological deficits scores, which include both neurological deficits and motor function, are commonly used in preclinical studies to measure stroke damage. In the MCAO/R model, infarct volume is an efficient indicator to evaluate whether the model is constructed successfully.23) In our experiment, the neuro-behavioral deficits were evaluated by the Zea–Longa scoring and the infarct volume was assessed by TTC. Results indicated that DA significantly ameliorated the neurological deficit scores and reduced infarct volumes among MCAO/R rats, indicating that DA represents a potent neuroprotective agent. This result is in agreement with the protective effect of DA against OGD/R injured PC12 cells in vitro.
Among the pathological events after stroke, inflammatory response and nerve cell damage are among the important factors affecting the recovery of stroke patients. tPA thrombolytic therapy, although being the main therapy for stroke, does not suppress neuro-inflammation and cannot directly recover the damaged nerve cells. Neuroprotective agents can exert a beneficial role in stroke therapy by reducing secondary injury to penumbra tissue or minimizing damage before and after reperfusion to promote neural recovery and plasticity.19,20) Our previous study has shown that DA could inhibit LPS-induced NO and proinflammatory cytokine production in BV2 microglia by suppressing NF-κB activation.22) The current study indicated that DA could exert a protective effect against OGD-damaged PC12 cells via suppressing the MKK7/JNK pathway. Further animal experiments also exhibited that DA could improve the neurological deficit scores and reduce the infarct volumes of MCAO/R rats. Therefore, DA could be a promising neuroprotective agent and will be helpful in the prevention and post-treatment of ischemic stroke.
CONCLUSION
Collectively, DA can significantly reduce apoptosis of PC12 cells caused by OGD/R, and the underlying mechanism might be related to suppressing the MKK7/JNK pathway. Furthermore, DA can protect MCAO/R-induced ischemic rats in vivo. These results showed that DA had the potential to be a neuroprotective agent and could be further studied as a lead compound for ischemic stroke treatment.
Acknowledgments
The work was supported by NSFC (82173704, 31870327, 82004215), The Key Research and Development Program of China (2017YFC1702002), National Major Project of China (2018ZX09731016-005), Shanghai Engineering Research Center for the Preparation of Bioactive Natural Products (16DZ2280200), Shanghai Municipal Health Commission Project (20204Y0326), Yunnan Provincial Science and Technology Department (202005AE160004, 202005AF150043, 2019HB025).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
REFERENCES
- 1) Herpich F, Rincon F. Management of acute ischemic stroke. Crit. Care Med., 48, 1654–1663 (2020).
- 2) Lapchak PA, Zhang JH. The high cost of stroke and stroke cytoprotection research. Transl. Stroke Res., 8, 307–317 (2017).
- 3) Powers WJ, Rabinstein AA, Ackerson T, Adeoye OM, Bambakidis NC, Becker K, Biller J, Brown M, Demaerschalk BM, Hoh B, Jauch EC, Kidwell CS, Leslie-Mazwi TM, Ovbiagele B, Scott PA, Sheth KN, Southerland AM, Summers DV, Tirschwell DL. Guidelines for the early management of patients with acute ischemic stroke: 2019 update to the 2018 guidelines for the early management of acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke, 50, e344–e418 (2019).
- 4) Wang X, Tsuji K, Lee SR, Ning M, Furie KL, Buchan AM, Lo EH. Mechanisms of hemorrhagic transformation after tissue plasminogen activator reperfusion therapy for ischemic stroke. Stroke, 35 (Suppl. 1), 2726–2730 (2004).
- 5) Paul S, Candelario-Jalil E. Emerging neuroprotective strategies for the treatment of ischemic stroke: an overview of clinical and preclinical studies. Exp. Neurol., 335, 113518 (2021).
- 6) Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, Moghaddam HF. Pathogenic mechanisms following ischemic stroke. Neurol. Sci., 38, 1167–1186 (2017).
- 7) Uzdensky AB. Apoptosis regulation in the penumbra after ischemic stroke: expression of pro- and antiapoptotic proteins. Apoptosis, 24, 687–702 (2019).
- 8) Sun J, Nan G. The mitogen-activated protein kinase (MAPK) signaling pathway as a discovery target in stroke. J. Mol. Neurosci., 59, 90–98 (2016).
- 9) Gao X, Li S, Liu X, Cong C, Zhao L, Liu H, Xu L. Neuroprotective effects of Tiaogeng decoction against H2O2-induced oxidative injury and apoptosis in PC12 cells via Nrf2 and JNK signaling pathways. J. Ethnopharmacol., 279, 114379 (2021).
- 10) Wang M, Hayashi H, Horinokita I, Asada M, Iwatani Y, Liu JX, Takagi N. Neuroprotective effects of Senkyunolide I against glutamate-induced cells death by attenuating JNK/caspase-3 activation and apoptosis. Biomed. Pharmacother., 140, 111696 (2021).
- 11) Yi X, Fan D, Yi T, Chen H, Qing T, Han Z, Bao S. 1-Trifluoromethoxyphenyl-3-(1-propionylpiperidin-4-yl) urea exerts neuro-protective effects against ischemic injury via suppressing JNK/p38 MAPK-mediated mitochondrial apoptosis pathway. J. Stroke Cerebrovasc. Dis., 30, 105957 (2021).
- 12) Liu H, Liu S, Tian X, Wang Q, Rao J, Wang Y, Xiang F, Zheng H, Xu L, Dong Z. Bexarotene attenuates focal cerebral ischemia-reperfusion injury via the suppression of JNK/caspase-3 signaling pathway. Neurochem. Res., 44, 2809–2820 (2019).
- 13) Xia P, Zhang F, Yuan Y, Chen C, Huang Y, Li L, Wang E, Guo Q, Ye Z. ALDH 2 conferred neuroprotection on cerebral ischemic injury by alleviating mitochondria-related apoptosis through JNK/caspase-3 signing pathway. Int. J. Biol. Sci., 16, 1303–1323 (2020).
- 14) Yang H, Liu Z, Liu X, Cao X, Chen M, Lou S, Rong L, Xu Y, Zhang Q. Tat-SynGAP improves angiogenesis and post-stroke recovery by inhibiting MST1/JNK signaling. Brain Res. Bull., 180, 38–45 (2022).
- 15) Toyoshima F, Moriguchi T, Nishida E. Fas induces cytoplasmic apoptotic responses and activation of the MKK7-JNK/SAPK and MKK6-p38 pathways independent of CPP32-like proteases. J. Cell Biol., 139, 1005–1015 (1997).
- 16) Sun Y, Lian M, Lin Y, Xu B, Li Y, Wen J, Chen D, Xu M, Almoiliqy M, Wang L. Role of p-MKK7 in myricetin-induced protection against intestinal ischemia/reperfusion injury. Pharmacol. Res., 129, 432–442 (2018).
- 17) Vercelli A, Biggi S, Sclip A, Repetto IE, Cimini S, Falleroni F, Tomasi S, Monti R, Tonna N, Morelli F, Grande V, Stravalaci M, Biasini E, Marin O, Bianco F, di Marino D, Borsello T. Exploring the role of MKK7 in excitotoxicity and cerebral ischemia: a novel pharmacological strategy against brain injury. Cell Death Dis., 6, e1854 (2015).
- 18) Tao T, Liu M, Chen M, Luo Y, Wang C, Xu T, Jiang Y, Guo Y, Zhang JH. Natural medicine in neuroprotection for ischemic stroke. Challenges and prospective. Pharmacol. Ther., 216, 107695 (2020).
- 19) Li SS, Lu YC, Ding DF, Ma ZZ, Xing XX, Hua XY, Xu JG. Fibroblast growth factor 2 contributes to the effect of salidroside on dendritic and synaptic plasticity after cerebral ischemia/reperfusion injury. Aging, 12, 10951–10968 (2020).
- 20) Xia DJ, Zhang Z, Zhao YL. Acteoside attenuates oxidative stress and neuronal apoptosis in rats with focal cerebral ischemia–reperfusion injury. Biol. Pharm. Bull., 41, 1645–1651 (2018).
- 21) Zhang ZY, Wang JX, Li J, Yang F, Liu GD, Tang WJ, He WW, Fu JJ, Shen YH, Li A, Zhang WD. Total synthesis and stereochemical assignment of delavatine A: Rh-catalyzed asymmetric hydrogenation of indenetype tetrasubstituted olefins and kinetic resolution through Pd-catalyzed triflamide-directed C-H olefination. J. Am. Chem. Soc., 139, 5558–5567 (2017).
- 22) Xie Q, Wu GZ, Yang N, Shen YH, Tang J, Zhang WD. Delavatine A, an unusual isoquinoline alkaloid exerts anti-inflammation on LPS-induced proinflammatory cytokines production by suppressing NF-kappaB activation in BV-2 microglia. Biochem. Biophys. Res. Commun., 502, 202–208 (2018).
- 23) Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke, 20, 84–91 (1989).
- 24) He Z, Ning N, Zhou Q, Khoshnam SE, Farzaneh M. Mitochondria as a therapeutic target for ischemic stroke. Free Radic. Biol. Med., 146, 45–58 (2020).
- 25) Liu F, Lu J, Manaenko A, Tang J, Hu Q. Mitochondria in ischemic stroke: new insight and implications. Aging Dis., 9, 924–937 (2018).
- 26) Lv S, Song Y, Zhang FL, Yan XL, Chen J, Gao L, Guo ZN, Yang Y. Early prediction of the 3-month outcome for individual acute ischemic stroke patients who received intravenous thrombolysis using the N2H3 nomogram model. Ther. Adv. Neurol. Disord., 13, 1756286420953054 (2020).
- 27) Feng L, Gao J, Liu Y, Shi J, Gong Q. Icariside II alleviates oxygen-glucose deprivation and reoxygenation-induced PC12 cell oxidative injury by activating Nrf2/SIRT3 signaling pathway. Biomed. Pharmacother., 103, 9–17 (2018).
- 28) Liu Y, Wu X, An J, Lv W, Geng Y, Lou T, Zhang Y. Glaucocalyxin B protects against oxygen-glucose-deprivation/reperfusion-induced neuronal injury in PC-12 cells. J. Cell. Biochem., 120, 6137–6144 (2019).
- 29) Zhang JF, Zhang L, Shi LL, Zhao ZH, Xu H, Liang F, Li HB, Zhao Y, Xu X, Yang K, Tian YF. Parthenolide attenuates cerebral ischemia/reperfusion injury via Akt/GSK-3beta pathway in PC12 cells. Biomed. Pharmacother., 89, 1159–1165 (2017).
- 30) Yang JL, Mukda S, Chen SD. Diverse roles of mitochondria in ischemic stroke. Redox Biol., 16, 263–275 (2018).
- 31) Su X, Zhou M, Li Y, Zhang J, An N, Yang F, Zhang G, Yuan C, Chen H, Wu H, Xing Y. Protective effects of natural products against myocardial ischemia/reperfusion: Mitochondria-targeted therapeutics. Biomed. Pharmacother., 149, 112893 (2022).
- 32) Orellana-Urzúa S, Rojas I, Libano L, Rodrigo R. Pathophysiology of ischemic stroke: role of oxidative stress. Curr. Pharm. Des., 26, 4246–4260 (2020).
- 33) Song X, Zhang L, Hui X, Sun X, Yang J, Wang J, Wu H, Wang X, Zheng Z, Che F, Wang G. Selenium-containing protein from selenium-enriched Spirulina platensis antagonizes oxygen glucose deprivation-induced neurotoxicity by inhibiting ROS-mediated oxidative damage through regulating MPTP opening. Pharm. Biol., 59, 629–638 (2021).
- 34) Li Y, Sun J, Wu R, Bai J, Hou Y, Zeng Y, Zhang Y, Wang X, Wang Z, Meng X. Mitochondrial MPTP: a novel target of ethnomedicine for stroke treatment by apoptosis inhibition. Front. Pharmacol., 11, 352 (2020).
- 35) Liao LX, Zhao MB, Dong X, Jiang Y, Zeng KW, Tu PF. TDB protects vascular endothelial cells against oxygen-glucose deprivation/reperfusion-induced injury by targeting miR-34a to increase Bcl-2 expression. Sci. Rep., 6, 37959 (2016).
- 36) Coffey ET. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci., 15, 285–299 (2014).
- 37) Centeno C, Repici M, Chatton JY, Riederer BM, Bonny C, Nicod P, Price M, Clarke PG, Papa S, Franzoso G, Borsello T. Role of the JNK pathway in NMDA-mediated excitotoxicity of cortical neurons. Cell Death Differ., 14, 240–253 (2007).
- 38) Song IS, Jun SY, Na HJ, Kim HT, Jung SY, Ha GH, Park YH, Long LZ, Yu DY, Kim JM, Kim JH, Ko JH, Kim CH, Kim NS. Inhibition of MKK7-JNK by the TOR signaling pathway regulator-like protein contributes to resistance of HCC cells to TRAIL-induced apoptosis. Gastroenterology, 143, 1341–1351 (2012).
- 39) Wu L, Zeng S, Cao Y, Huang Z, Liu S, Peng H, Zhi C, Ma S, Hu K, Yuan Z. Inhibition of HDAC4 attenuated JNK/c-Jun-dependent neuronal apoptosis and early brain injury following subarachnoid hemorrhage by transcriptionally suppressing MKK7. Front. Cell. Neurosci., 13, 468 (2019).
- 40) Li Y, Zhang J. Animal models of stroke. Animal Model. Exp. Med., 4, 204–219 (2021).
- 41) Li K, Ding D, Zhang M. Neuroprotection of osthole against cerebral ischemia/reperfusion injury through an anti-apoptotic pathway in Rats. Biol. Pharm. Bull., 39, 336–342 (2016).