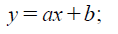Abstract
Ginkgolide B (GKB) is a well-established neuroprotectant for acute ischemia stroke. However, its cerebral exposure and real-time response remain elusive in acute ischemia/reperfusion stage, and it hinders its usage in therapeutic window of ischemia stroke. Therefore, we investigate the exposure-response relationship of GKB (10 mg/kg, intravenously (i.v.)) as well as its neuroprotective mechanism in acute ischemia/reperfusion rats. Cerebral and plasma exposure of GKB is comparatively explored in both of normal rats and acute ischemia/reperfusion rats. Correspondingly, neurological function and brain jury indexes were assessed at each time point, and superoxide dismutase (SOD), malondialdehyde (MDA), platelet activator factor (PAF) and thromboxane A2 (TXA2) are indexed as pharmacological response to GKB. Exposure-response relationships are analyzed by using linear regression. Additionally, cerebral expressions of proteins in PAF-regulated pathways are tested at each time point. Results show cerebral and plasma concentrations of GKB are much higher in acute ischemia/reperfusion rats than those in normal rats. Cerebral infarction, neurological function (NF) score, abnormal PAF and excessive MDA are significantly alleviated in 24 h after GKB injection, and PAF is reduced in exposure-response manner with significant concentration–response relationship (R2 = 0.9123). Regarding downstream proteins in intracellular PAF-regulated pathway, GKB progressively inhibits Bax, Caspase-3, p-p65 and p-IKK, while gradually restoring LC3B, p62 and p-mammalian target of rapamycin (mTOR) to the basic level within 24 h. Conclusively, GKB exhibits greater cerebral exposure in acute ischemia/reperfusion rats and neuroprotective effect through reducing PAF in exposure-response manner and mediating PAF-regulated intracellular signaling pathways. Our finding highlights clinical implications of GKB in therapeutic time window of ischemic stroke.
INTRODUCTION
Stroke is listed as second leading cause of death and disability worldwide1) and its occurrence is highest among fatal diseases in China.2) Ischemic stroke, which is a major subtype of stroke and accounts for 80% of stroke,1) needs a long-term medical care from neuronal injury,3) which may be associated with timing treatment in the acute stage. To minimize the neuronal damage in ischemia stroke, it is critical to conduct recanalization timely within 4.5 h of ischemia stroke onset.4) To date, various recanalization treatments are available for ischemia stroke, however, ischemia/reperfusion-induced neurotoxicity is inevitable in current treatments,5) and it leads to irreversible neuron damage and even disability.6) Therefore, immediate neuroprotection is necessarily initiated from onset of acute ischemia stroke in order to minimize nervous damage of acute ischemia/reperfusion. Despite of great investment, numerous neuroprotectant candidates cannot prove to be beneficial in clinic. Recently, plant-derived compounds are revealed to be potential neuroprotectants for ischemia stroke,7) but they still need more accurate investigation for clinic application.
Ginkgolide B (GKB) is one of natural compounds derived from Ginkgo biloba L., which has long been used as traditional neuroprotectant in China.8) Its neuroprotective effect has attracted numerous attentions from research and clinical practitioners.9–11) However, its neuroprotective effect was evaluated at a final endpoint instead of dynamic monitor in acute phage of ischemia stroke,12–17) and current findings are not convincing enough for application of GKB in emergency of acute ischemia stroke. Exposure-response analysis is a powerful tool to evaluate relationship between concentration of agents and its real-time pharmacological regulation,18) and it can further elucidate rationality of GKB treatment in acute ischemia stroke.
In order to evaluate GKB’s real-time efficacy and rationality in acute ischemia/reperfusion stage, exposure-response analysis and hourtime-depended mechanism experiment is conducted in middle cerebral artery occlusion (MCAO)-induced ischemia-stroke model in time period of 2–6 h after GKB administration. In regard to response index, superoxide dismutase (SOD), malondialdehyde (MDA), platelet activator factor (PAF) and thromboxane A2 (TXA2), are chosen since they are representative biomarkers of ischemia/reperfusion injury. Noticeably, platelet activating factor (PAF) is a potent phospholipid promoting inflammation and platelet aggregation, and excessive PAF is a feature of ischemia stroke and causes inflammation and apoptosis in both immunocyte and neurocytes. Moreover, ischemic brain injury can be prevented by inhibiting PAF, which can be antagonized by GKB.19) Therefore, PAF is considered as a critical index of GKB for protecting brain tissue in ischemia/reperfusion. However, little is known about its dynamic alternation of PAF and related pathways after GKB intervention in time window. Therefore, hourtime-depenedent regulation on PAF-mediating targets, including p-IKK and p-p65 (inflammation), Bcl-2, Bax and Caspase-3 (apoptosis), and p-mammalian target of rapamycin (mTOR), LC3, p62 (autophagy), are explored for understanding mechanism of GKB’s neuroprotective effect. Our research provides further insight into exposure-response relationship of GKB in acute stage of ischemia stroke.
MATERIALS AND METHODS
MCAO ModelA total of 216 male Sqrague–Damley (SD) rats weighing 320–340 g were purchased from the Guangdong Medical Laboratory Animal Center (Lot No. 44007200044705). Rats were randomly divided into 2 groups in pharmacokinetic experiment, including GKB in normal group (n = 6 for each time point) and GKB in MCAO (n = 6 for each time point). Rats were randomly divided into 3 groups in response experiment, including normal group (n = 6 for each time point), MCAO model group (MCAO group without GKB treatment, n = 6 for each time point) and GKB group (MCAO group with GKB treatment, n = 6 for each time point). Rats were anesthetized with isoflurane (5% induction and 2% maintenance) in a calibrated vaporizer, and received MCAO surgery (MCAO group) or surgery without MCAO (sham group) as described previously20) with minor modification (The filament was left in place for 120 min after which it was withdrawn from the internal carotid artery to allow reperfusion.). Reperfusion time is initiated 2 h after occlusion, and single dose of 10 mg/kg Ginkgolinde B (provided by Selleckchem, U.S.A., purity ≥ 99%) was intravenously (i.v.) injected 5 min after reperfusion. Experiment was performed according to the Guidelines of Sun Yat-sen University, Sun Yat-sen University Animal Ethics Committee. The experimental protocols were approved by Sun Yat-sen University Animal Ethics Committee. All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
Neurological Function Scores (NFs)Neurological scoring tests were carried out at different time points (2, 4, 6, 24 h) after MCAO. The assessment consisted of 6 repeats and was divided by 0–4 point scale: No observed neurological deficits—0 score; failed to outstretch left forelimb (mild focal neurological impairment)—1 score; Turn left while walking (moderate focal neurological impairment)—2 score; Lean to the left (severe focal neurological impairment)—3 score; Unable to walk spontaneously—4 score. The test was performed by a blinded investigator.
Cerebral Index Measurement and Cerebral Infarction RateAll brains were quickly harvested after the rats were sacrificed and washed with 4 °C normal saline for 3 times, dehydrated in filter paper for 2 min and weighed.
Brain index = brain weight (mg)/body weight (g). The obtained brains were coronally sliced into five sections (2.00 mm-thick). After the presence of intracranial hemorrhage visible to the naked eye, brain slices were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) for 30 min at 37 °C in dark. The prepared samples were photographed and the infarct areas were measured using Image-Pro Plus 6.0. The cerebral infarction rate refers to the percentage of infarct volume in the ipsilateral hemisphere of the lesion.
Determination of SOD, MDA, PAF and TXA2 in the Brain TissuesRats were sacrificed under deep anesthesia and the brains were harvested at each time point after GKB administration. Brain tissue (about 0.1 g) was homogenized in cold phosphate buffered saline (PBS) of 10-fold volume to tissue weight (mL/mg) on ice. The homogenate was immediately centrifuged at 10000 rpm for 10 min, and then the supernatant was collected for determination of SOD, MDA, PAF and TXA2 by using kits (Nanjing Jiancheng Bioengineering Institute) under the manufacturer’s instructions.
Determine the Content of GKB in Plasma and Brain TissuesFor plasm sample preparation, 10 μL ginkgolide C (GKC) internal standard solution (20 μg/mL) and 2 μL HCl solution (2.5 mol/L) were added into a 100 μL plasma sample. The mixture was extracted with 1 mL ethyl acetate by vortex-mixing for 2 min, and was then centrifugate at 13000 rpm for 5 min. The supernatant (900 μL) was transferred to a new 1.5 mL Eppendorf tube and blow-dried by nitrogen at 40 °C. The residue was resuspended with 200 μL methanol-water (40 : 60), then centrifugation at 13000 rpm for 10 min again. The supernatant (1 μL) was injected for ultra performance liquid chromatography (UPLC)-MS analysis.
For brain sample preparation, brain tissues were cut into pieces, then 1 g tissue was homogenized with 3 mL saline solution of 50 mM HCl. After centrifugation at 13000 rpm for 5 min, supernatant (5 μL) was injected for UPLC-MS analysis.
Samples were analyzed by Waters Alliance® 2695 with 2996 PDA and Micromass® Quattro microTM using ACQUITY UPLCBEH® C18 column (2.1 × 50 mm, 1.7 µm) with an elution program consisting of 0.1% aqueous formic acid (A)-menthol (B) (60 : 40) at a flow rate of 0.3 mL/min. Detection was performed under the selected reaction monitoring (SRM) scan using an electrospray ionization (ESI) in the positive ion mode. The mass transitions were as follows: m/z 423.1→367.1 for GKB, and m/z 439.1→383.1 for GKC (Inner Standard). The MS conditions were as follows: Spray Voltage, 3.0 kV; Vaporizer Temperature, 200 °C; Capillary Temperature, 270 °C; Sheath Gas Pressure, 30 psi; Aux Gas Pressure, 10 psi. Data collection and processing were conducted with winnolin 7.0.
Western Blot AnalysisThe left hemisphere (MCAO) and right hemisphere (intact) of brain tissues (approximately 100 mg) were respectively homogenized in 1 mL ice-cold radio immunoprecipitation assay (RIPA) buffer with 1% protease inhibitor cocktail (BestBio, Shanghai, China). Western blot assay was performed as previously described with modification.18) Primary antibodies were used at dilutions as follow: anti-Bcl-2 (1 : 500, Abcam, U.K.), anti-Bax (1 : 2000, Abcam), anti-cleaved-caspase-3 (1 : 500, CST), anti-phosphorus-IKKα/β (1 : 1000, CST), anti-phosphorus-nuclear factor-kappaB (NFκB) (1 : 1000, CST), anti-phosphorus-mTOR (1 : 1000, Abcam), anti-p62 (1 : 2000, Abcam), anti-LC3B (1 : 1000, Abcam), anti-β-actin (1 : 5000, Sigma). The bands were visualized by means of enhanced chemiluminescence. The exposure was performed with 5220 Multi (Tanon, Shanghai, China), and the acquired image was analyzed with Tanon Gis (Tanon). Blot bands were quantified by densitometry using ImageJ software. Relative level of protein expression level in left hemisphere (MCAO) is normalized by counterpart in right hemisphere (intact).
Statistical AnalysisSPSS 22.0 was used for statistical analysis. The results were presented as mean ± standard deviation (x̅ ± S.D.) from. Bilateral t test or one-way ANOVA were used to analyze neurological scores. Neurological scores between groups were compared the Kruskal–Wallis test followed by the Mann–Whitney U test. p < 0.05 was considered statistically significant.
In order to analyze the response efficiency of GKB, Area under curve (AUC), ᶿAUC and Response Efficiency are applied as parameters for analyzing real-time efficacy of GKB on response index in time-response experiment.
t: time after administration; Y: index value
Thus, linear regression model is applied to analyze the relationship between exposure and response efficiency, the equation is shown and described as follow:
-
y: RE, a negative percentage representing reduced percent of response index. It includes 4 y values: REt2-t3, REt3-t4, REt4-t6 and REt6-t24 for each index.
-
x: concentration of GKB. It includes 4 x values: Ct3, Ct4, Ct6 and Ct24 for each index.
-
a: slope. If it is negative number, it shows positive relationship between y (RE) and x (GKB concentration in each time point; If it is positive number, it shows negative relationship.
RESULTS
Plasma and Intracranial Concentration of GKB in MCAO-Induced Ischemia Stroke RatsAs showed in Fig. 1A, compared with sham group, elimination of plasma GKB was prolonged in MCAO rats and concentration of GKB is much higher in MCAO. In 2, 3, 4 h time point after i.v. injection of GKB, plasma concentration of GKB is much greater than that in normal group. At these three time points, the concentration of GKB kept at high level (537.92 ± 165.84 ng/mL in 2 h, 520.90 ± 168.30 ng/mL in 3 h, and 480.58 ± 222.66 ng/mL in 6 h) while that of normal group displayed remarkable decrease from 396.32 ± 52.83 ng/mL (2 h) to 134.09 ± 59.10 ng/mL (4 h). Notably, it remains 35.27 ± 28.06 ng/mL 24 h after GKB treatment in MCAO group, whereas it cannot be detected in normal group.
As shown in Fig. 1B, intracranial distribution of GKB is significantly higher in MCAO group than counterpart in normal group from 3 to 24 h after GKB treatment, and intracranial elimination of GKB shows much slower compared with normal group. In 6 h and 24 time points, the concentration of GKB remains retained 8.33 ± 7.21 ng/mL and 5.01 ± 4.74 ng/mL respectively, while it cannot be detected in normal group 6 h after the injection.
GKB Attenuates Cerebral Infarction in Exposure-Response Manner in MCAO-Induced Ischemia Stroke RatCerebral infarction rate was analyzed in exposure-response linear model. As shown in Figs. 2 and 3A, cerebral infarction is significantly attenuated by GKB in 24 h, and this attenuation is triggered in 2–6 h after ischemia/reperfusion. Relationship between response efficiency (RE = ᶿAUC/AUCMCAO) and GKB exposure is investigated. There was mild linear relationship (y for response efficiency, x for plasma concentration of GKB; y = − 0.1100x − 23.80, R2 = 0.7191, see in Fig. 3B) between plasma concentration of GKB and response efficiency (RE) within 24 h, and notably similar relationship (y for response efficiency (RE), x for intracranial concentration of GKB; y = − 4.423x − 6.567, R2 = 0.7542, see in Fig. 3C) between cerebral concentration of GKB and response efficiency (RE) in cerebral infarction. Taken together, these outcomes indicate that positive and mild relationship is observed in GKB exposure and cerebral infraction index, but more precise index is needed to explore exposure-response of GKB.
GKB Improves Neurological Function in Mild Exposure-Response Manner in MCAO-Induced Ischemia Stroke Rat, but Does Not Affect Brain IndexNeurological deficits were applied to evaluate neurological function in MCAO-induced ischemia stroke rat treated with GKB for 2, 3, 4, 6, 24 h. As shown in Fig. 4A, GKB remarkably reduced Neurological deficits score in all time points. After GKB treatment, Response Efficiency of neurological deficits score shows mild linear relationship with cerebral concentration of GKB (y = − 0.4047x − 23.23, R2 = 0.7793, see Fig. 5B), and positive relationship was observed with plasma concentration of GKB (y = − 0.0099x − 24.87, R2 = 0.7159, see Fig. 4B). Additionally, brain index is not affected by GKB exposure (Figs. 5A, B, C).
GKB Reduced PAF in Remarkable Exposure-Response Manner in MCAO-Induced Ischemia Stroke Rats, but Mildly Eliminated MDA and Hardly Affected TXA2 and SODI/R injury indexes, including PAF, TXA2, MDA, and SOD, were employed to evaluate neuroprotection of GKB against MCAO-induced ischemia stroke. As Fig. 6A shows, PAF was significantly reduced PAF in 2–6 h after GKB injection (p < 0.05). However, MDA, TXA2 and SOD are not affected by GKB exposure as Figs. 7A, 8A, and 8B show. After GKB treatment, reduced PAF presents significant linear relationship with cerebral exposure of GKB (y = − 1.635x − 19.79, R2 = 0.9123, Fig. 6B), and positive relationship was observed with concentration of GKB in plasma (y = − 0.0214x − 25.93, R2 = 0.9013, Fig. 6A). Specifically, it achieves greatest response of eliminating PAF at 2 and 3 h time points, in which intracranial concentration of GKB is in its peak. Mild relationship between GKB exposure and reduced percentage of MDA is observed in this research (plasma: y = − 0.0260x − 19.75, R2 = 0.6995; cerebral: y = − 0.9741x − 196.52, R2 = 0.6995).
GKB Regulates Protein Expressions of Ischemia/Reperfusion-Induced Inflammation, Apoptosis and Autophagy in Brain TissueIn term of inflammation (Figs. 9A, B), MCAO-induced p-IKKα/β and p-p65 are remarkably reversed to normal levels 6 and 24 h after exposure of GKB, while these proteins are hardly affected by higher cerebral concentration of GKB at 2 and 4 h time points. In term of apoptosis (Figs. 9C–E), time-depended increase of Caspase-3 and Bax is observed in MCAO group while anti-apoptosis protein Bcl-2 is inhibited within 24 h in MCAO group. After treatment of GKB, restoration of GKB on caspase-3, bax and Bcl-2 is sustainable even in 24 h. In term of autophagy, mTOR, an autophagy suppressor, is inhibited by MCAO, and LC3B, index of autophagy activation, is increased by MCAO (Figs. 9F, G). Additionally, p62, recruit protein, is inhibited after MCAO surgery (Fig. 9H). After treatment with GKB, LC3B is inhibited in 2 and 4 h, and it is normalized at 24 h in neuron (Fig. 9G). p-mTOR is gradually inhibited by GKB treatment when compared with model group (Fig. 9F). However, p62 is restored to normal level in 4 h and 6 h in rats treated with GKB, but its expression is significantly upregulated by GKB in 24 h (Fig. 9H).
DISCUSSION
Ischemia reperfusion (I/R)-induced neuron damage is a major cause of disability in ischemia stroke patients, but this irreversible injury remains inevitable in current treatments. Noticeably, some neuroprotective agents are beneficial for relieving I/R injury and promoting recovery, but they are not included in clinical practice due to unclarified effect and mechanism in therapeutic time window. Therefore, in our research, exposure-response analysis and dynamic action mechanism are investigated within therapeutic time window. In other words, the core of our research is to confirm whether Ginkgolide B (GKB) possesses quick cerebral exposure and fast-acting property on neuroprotection in therapeutic time window. The most significant finding in our research is that cerebral exposure of GKB is enhanced in MCAO (middle cerebral artery occlusion)-induced ischemia rats, and GKB exerts profound neuroprotective effect against MCAO-induced acute ischemia stroke through reducing cerebral PAF in exposure-response manner and regulating PAF-downstream pathways: inhibiting Bcl-2/Caspase-3 apoptosis and IKK/NFκB inflammation pathways, and equilibrating disordered autophagy (shown in Fig. 10).
GKB Reduces PAF and Exerts Neuroprotection in Exposure-Response Manner, and Its Neuroprotective Effect Lasts in Post-exposure PeriodIn term of GKB exposure, it is previously reported that brain tissue distribution of GKB is increased in MCAO rats,21) and our results supported this contention. Specifically, cerebral distribution of GKB is remarkably enhanced after occlusion-induced ischemia and reperfusion in our case. Furthermore, underlying mechanism of enhanced GKB permeate is documented to be related to disrupted tight junctions (TJs), which was caused by hypoxia and reoxygenation in ischemia/reperfusion.21) In other words, cerebral GKB exposure is increased due to Blood–Brain-Barrier (BBB) damage caused by MCAO. As a consequence of breakdown of BBB, distribution and elimination of GKB are simultaneous in both plasma and brain tissue. Therefore, ischemia/reperfusion-induced pathophysiological alternation contribute to cerebral exposure of agents including GKB, but it is not exclusive that other factors may involve in phenomenon. In our case, excessive PAF may serve as a factor for promoting exposure of GKB since interaction between GKB and PAF may prolong elimination of GKB or change distribution. Therefore, effect of PAF on GKB’s distribution is undergone in our lab but it is inclusive yet. GKB is metabolized by hydrolysis in plasma and is eliminated primarily through urinary excretion (approx.48%). Plus, GKB is also a potential substrate for organic anion transporters and/or organic anion transporting polypeptides (OATs/OATPs) according to the previous research.22) In this regard, GKB metabolism can be prolonged by inhibition of hydrolase and transporter. In the condition of ischemia/reperfusion, the excessive PAF in plasma or organ may serve as an endogenous molecule to affect GKB’s metabolism enzymes or transporters in plasma, brain and kidney since the majority of PAF was hydrolyzed in liver and kidney through PAF-AH,23) which is hydrolase for PAF.24) Therefore, it is possible that PAF prolonged its elimination through mechanistical interaction or interrupting hydrolysis of GKB and transport in kidney. Additionally, intracellular PAF is antagonist by GKB and vice versa, and, therefore, PAF will prolong elimination of GKB as a competitive substrate of intracellular PAFR or PLA2G7 (PAF-AH), which are target of GKB as well.25) In sum, quick GKB distribution is made between plasma and brain tissue within 24 h and it shows rational to utilize GKB in ischemia stroke emergency regarding cerebral exposure.
In term of response research, endogenous thrombin and lipid peroxidation are indexed for evaluating neuroprotective effect of GKB since I/R injury is defined as secondary injury caused by pathogenesis from vascular occlusion, including high endogenous thrombin potential26) and oxidative product. Platelet activating factor (PAF, 1-O-alkyl-2-acetyl-snglycero-3-phosphocholine) is a major lipid mediator response for neurotoxicity27,28) in acute stage of I/R injury. It accumulates in brain tissue and triggers inflammation and generation of reactive oxygen species (ROS) in neuron through PAF-PAF receptor pathway and PAF-neurotransmitter interaction after ischemia stroke.29,30) Thromboxane A2 (TXA2) is an intermediate metabolized from arachidonic acid,31) and it can impair circulatory function and aggravates injury through causing vascular contraction and endothelium dysfunction during ischemic stroke.32) MDA and SOD, which are two indexes for oxidation stress, can be regulated by PAF and TXA2 in ischemia stroke. In our research, PAF reduction is found to be a unique index with significant relationship with GKB exposure. By contrast, level of SOD is not affected by GKB exposure in 10 mg/kg dosage. Taken together, short-term exposure of GKB has preference on targeting PAF rather than activating anti-oxidation enzyme in 10 mg/kg dosage. Therefore, primary PAF-targeting property of GKB is responsive for its neuroprotective effect in 10 mg/kg dosage. Although GKB possesses various regulations on coagulation and oxidative stress in different exposure level,33) it exhibits PAF-reducing effect without restoring oxidative stress in dosage of 10 mg/kg in our case. Thus, these findings further confirm that GKB has advantage for its usage in acute ischemia stroke through selectively eliminating PAF-dependent I/R injury within therapeutic window, and therapy window of GKB for PAF-targeting needs more investigation. Furthermore, although PAF-reducing response is greatly associated with concentration of GKB, it is still not exclusive that other mechanism such as cellular defensive is involved in GKB’s neuroprotection in concentration-independent manner in post-exposure of GKB.
Neuroprotective Effect of GKB Is Associated with Prolonged Mediation on Intracellular PAF-Regulated Inflammation, Apoptosis and Autophagy PathwaysInflammation and cell apoptosis, which cause neuron injury, can be triggered through PAF-induced extracellular signaling pathway28) and worsened by disordered autophagy under various stress. In context of I/R injury, IKK/NFκB inflammation and Bcl-2/Bax/Caspase-3 apoptosis represents downstream of PAF-induced injury, and mTOR/p62 autophagy represents defensive mechanism against injury. PAF-related pathways and autophagy can be simultaneously regulated after GKB treatment according to previous research,34) and our findings confirm GKB’s regulation on PAF-downstream pathway and autophagy restoration. Moreover, our results indicate dynamic regulations of inflammation and autophagy are partially independent of PAF reduction. To sum up, neuroprotective mechanism of GKB attributes to both PAF-reducing effect and prolonged effect on inhibiting inflammation and apoptosis and normalizing autophagy under various stress.
NF-κB is a nuclear factor which ubiquitously regulates inflammation response and cell-survival in neuron and immunocytes through IKKα/β pathways.35) Activation of NF-κB is reported to play a critical role in ischemia/reperfusion-induced brain injury.35) In case of ischemia/reperfusion, PAF severs as a stimulus in IKK/NFκB pathway activation. In our research, PAF and NFκB are efficiently decreased by high level of GKB exposure while IKK/ NFκB but not PAF is efficiently suppressed by GKB of low level. According to previous research,36) cellular defensive mechanism (autophagy) may be responsive for latter phenomenon, and it may be triggered to counteract the inflammation in the downhill GKB exposure.
Bcl-2 and Bax are two critical enzymes regulating cell apoptosis. Bcl-2 serves as an anti-apoptosis enzyme while Bax serves as a pro-apoptosis enzyme in cerebral ischemic injury.37) Additionally, Caspase-3 is a marker of apoptosis, and it can coordinate downstream signaling from Bcl-2/Bax complex to execute cell death program. In our research, upregulation of Bax protein and downregulation of Bcl-2 are observed within 24 h after MCAO-induced ischemia/reperfusion, showing the consistence with previous finding.38) Moreover, GKB-induced anti-apoptosis effect (Bcl-2-upregualtion, Bax-downregulation and caspase-3 inhibition) are observed in early and late stage in elimination of GKB, indicating that anti-apoptosis effect is prolonged from early-exposure to post-exposure periods of GKB. In early exposure stage of GKB, GKB-lead PAF reduction may attribute to anti-apoptosis due to that PAF reduction alleviates cell apoptosis in I/R situation. In late stage of GKB exposure, restoration of cellular defensive mechanism may contribute to neuroprotection of GKB.
Autophagy plays a critical role in neuron protection, which includes cell survival maintaining, toxic elimination and recycle of impaired organelles. Autophagy regulation is considered to be a protective procedure in neuron against various stresses,39) and it is also a regulative target of tissue-type plasminogen activator (tPA) for ischemia stroke.40) Furthermore, activation of autophagy can reduce inflammation or apoptosis in neuron in condition of ischemia/reperfusion while excessive autophagy is reported to cause cell death in cerebral ischemia,41,42) therefore, suitable regulation is needed in regard of pharmacological intervention on autophagy. I/R-induced dysfunction of autophagy is observed in our research, and inflammation and cell apoptosis may be associated with disorder autophagy according to previous findings.43,44) Furthermore, GKB demonstrates equilibrating effect on autophagic proteins which are responsive for various stages of autophagy in our research. GKB stabilizes autophagy initiation marker LC3B to basic level through a long exposure time, inhibits activation of autophagy regulator mTOR and increase expression of autophagosome recruit protein P62.45) Since p62 may be key enzyme for turnover of autophagic function through connecting pro-apoptosis or anti-apoptosis proteins,46,47) its expression reflects not only regulation of GKB on autophagy but also cell death and inflammation of neurocytes. Our findings indicates that regulation of p62 is associated with GKB-equilibrating autophagy, but more in-depth research is needed. Therefore, autophagy regulation is prospective to serve as complementary therapeutic target to our exposure-response findings.
Collectively, our findings indicate that lasting neuroprotection of GKB is not completely concentration-dependent, and it may be associated with trigger of protective mechanism against PAF-induced intracellular injury and turnover of harmful autophagy into prosthetic one in neuron. However, our finding is not conclusive due to limitation in mechanism research. Especially, autophagy regulation of GKB is still preliminary due to uncertainty on its effect in immune cell regulation in brain48) such as tissue repair function of microglial,12) which plays a crucial role in neuroprotection.49)
CONCLUSION
Our research firstly revealed that GKB exerts profound neuroprotective effect and reduces cytotoxic intermediates against MCAO-induced acute ischemia stroke through reducing PAF accumulation in exposure-response manner and regulating intracellular apoptosis, inflammation and autophagy pathways. Prolonging effect of GKB-triggered autophagy may play an important in elucidating its prolonging neuroprotective effect, and this finding will be novel insight into exploring neuroprotectant for ischemia stroke. Our finding indicates that GKB can serve as potential neuroprotectant from onset of ischemia reperfusion injury.
Acknowledgments
The authors appreciate the financial supports provided by National Key Research and Development Program of China during the 13th five year plan [No. 2018ZX09734-003]; the National Key Research and Development Program [No. 2017YFC0909303]; Guangdong Provincial Science and Technology Project [No. 2017B020234005]; the Guangdong Science and Technology Planning Project [No. 2016A040403047]; Guangdong Natural Science Foundation [No. 2015A030310230]; the National Key Research and Development Program [No. 2016YFC0905001] and Guangdong Provincial Key Laboratory of Construction Foundation [No. 2017B030314030].
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Harpaz D, Eltzov E, Seet RCS, Marks RS, Tok AIY. Point-of-care-testing in acute stroke management: an unmet need ripe for technological harvest. Biosensors, 7, 30 (2017).
- 2) Wu S, Wu B, Liu M, et al. Stroke in China: advances and challenges in epidemiology, prevention, and management. Lancet Neurol., 18, 394–405 (2019).
- 3) Su ZF, Sun ZW, Zhang Y, Wang S, Yu QG, Wu ZB. Regulatory effects of miR-146a/b on the function of endothelial progenitor cells in acute ischemic stroke in mice. Kaohsiung J. Med. Sci., 33, 369–378 (2017).
- 4) Kim JS. tPA helpers in the treatment of acute ischemic stroke: are they ready for clinical use? Journal of Stroke, 21, 160–174 (2019).
- 5) Chen HS, Qi SH, Shen JG. One-compound-multi-target: combination prospect of natural compounds with thrombolytic therapy in acute ischemic stroke. Curr. Neuropharmacol., 15, 134–156 (2017).
- 6) Uphaus T, Bittner S, Groschel S, Steffen F, Muthuraman M, Wasser K, Weber-Kruger M, Zipp F, Wachter R, Groschel K. NfL (neurofilament light chain) levels as a predictive marker for long-term outcome after ischemic stroke. Stroke, 50, 3077–3084 (2019).
- 7) Shang YH, Tian JF, Hou M, Xu XY. Progress on the protective effect of compounds from natural medicines on cerebral ischemia. Chin. J. Nat. Med., 11, 588–595 (2013).
- 8) Birks J, Grimley Evans J. Ginkgo biloba for cognitive impairment and dementia. Cochrane Database Syst. Rev., 20, CD003120 (2009).
- 9) Al-Adwani DG, Renno WM, Orabi KY. Neurotherapeutic effects of Ginkgo biloba extract and its terpene trilactone, ginkgolide B, on sciatic crush injury model: a new evidence. PLOS ONE, 14, e0226626 (2019).
- 10) Yin JJ, He Y, An J, Miao Q, Sui RX, Wang Q, Yu JZ, Xiao BG, Ma CG. Dynamic balance of microglia and astrocytes involved in the remyelinating effect of ginkgolide B. Front. Cell. Neurosci., 13, 572 (2020).
- 11) Feng Z, Sun Q, Chen W, Bai Y, Hu D, Xie X. The neuroprotective mechanisms of ginkgolides and bilobalide in cerebral ischemic injury: a literature review. Mol. Med., 25, 57 (2019).
- 12) Chen A, Xu Y, Yuan J. Ginkgolide B ameliorates NLRP3 inflammasome activation after hypoxic-ischemic brain injury in the neonatal male rat. Int. J. Dev. Neurosci., 69, 106–111 (2018).
- 13) Guo C, Zhang J, Zhang P, Si A, Zhang Z, Zhao L, Lv F, Zhao G. Ginkgolide B ameliorates myocardial ischemia reperfusion injury in rats via inhibiting endoplasmic reticulum stress. Drug Design. Devel. and Ther., 13, 767–774 (2019).
- 14) Liu Q, Jin Z, Xu Z, Yang H, Li L, Li G, Li F, Gu S, Zong S, Zhou J, Cao L, Wang Z, Xiao W. Antioxidant effects of ginkgolides and bilobalide against cerebral ischemia injury by activating the Akt/Nrf2 pathway in vitro and in vivo. Cell Stress Chaperones, 24, 441–452 (2019).
- 15) Yang X, Zheng T, Hong H, Cai N, Zhou X, Sun C, Wu L, Liu S, Zhao Y, Zhu L, Fan M, Zhou X, JSin F. Neuroprotective effects of Ginkgo biloba extract and Ginkgolide B against oxygen-glucose deprivation/reoxygenation and glucose injury in a new in vitro multicellular network model. Front. Med., 12, 307–318 (2018).
- 16) Zheng PD, Mungur R, Zhou HJ, Hassan M, Jiang SN, Zheng JS. Ginkgolide B promotes the proliferation and differentiation of neural stem cells following cerebral ischemia/reperfusion injury, both in vivo and in vitro. Neural Regen. Res., 13, 1204–1211 (2018).
- 17) Zheng Y, Wu Z, Yi F, Orange M, Yao M, Yang B, Liu J, Zhu H. By activating Akt/eNOS bilobalide B inhibits autophagy and promotes angiogenesis following focal cerebral ischemia reperfusion. Cell. Physiol. Biochem., 47, 604–616 (2018).
- 18) Clout AE, Della Pasqua O, Hanna MG, Orlu M, Pitceathly RDS. Drug repurposing in neurological diseases: an integrated approach to reduce trial and error. J. Neurol. Neurosurg. Psychiatry, 90, 1270–1275 (2019).
- 19) Zimmerman GA, McIntyre TM, Prescott SM, Stafforini DM. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit. Care Med., 30 (Suppl.), S294–S301 (2002).
- 20) Domin H, Przykaza L, Jantas D, Kozniewska E, Boguszewski PM, Smialowska M. Neuroprotective potential of the group III mGlu receptor agonist ACPT-I in animal models of ischemic stroke: in vitro and in vivo studies. Neuropharmacology, 102, 276–294 (2016).
- 21) Fang W, Deng Y, Li Y, Shang E, Fang F, Lv P, Bai L, Qi Y, Yan F, Mao L. Blood brain barrier permeability and therapeutic time window of Ginkgolide B in ischemia-reperfusion injury. Eur. J. Pharm. Sci., 39, 8–14 (2010).
- 22) Shen C, Jin X, Wu M, Huang X, Li J, Huang H, Li F, Liu J, Rong G, Song S. A sensitive LC-MS/MS method to determine ginkgolide B in human plasma and urine: application in a pharmacokinetics and excretion study of healthy Chinese subjects. Xenobiotica, 50, 323–331 (2020).
- 23) Liu J, Chen R, Marathe GK, Febbraio M, Zou W, McIntyre TM. Circulating platelet-activating factor is primarily cleared by transport, not intravascular hydrolysis by lipoprotein-associated phospholipase A2/ PAF acetylhydrolase. Circ. Res., 108, 469–477 (2011).
- 24) Nakamura Y, Yoshida M, Tanigawa K, Harada A, Kihara-Negishi F, Maruyama K, Karasawa K. Deficiency of type I platelet-activating factor-acetylhydrolase catalytic subunits causes an increase in body weight. Biol. Pharm. Bull., 44, 920–925 (2021).
- 25) Ling SK, Pisar MM, Man S. Platelet-activating factor (PAF) receptor binding antagonist activity of the methanol extracts and isolated flavonoids from Chromolaena odorata (L.) King and Robinson. Biol. Pharm. Bull., 30, 1150–1152 (2007).
- 26) Goldman S, Prior SM, Bembenek JP, Niewada M, Broniatowska E, Czlonkowska A, Butenas S, Undas A. Activation of blood coagulation and thrombin generation in acute ischemic stroke treated with rtPA. J. Thromb. Thrombolysis, 44, 362–370 (2017).
- 27) Sirangelo I, Giovane A, Maritato R, D’Onofrio N, Iannuzzi C, Giordano A, Irace G, Balestrieri ML. Platelet-activating factor mediates the cytotoxicity induced by W7FW14F apomyoglobin amyloid aggregates in neuroblastoma cells. J. Cell. Biochem., 115, 2116–2122 (2014).
- 28) Ryan SD, Harris CS, Mo F, Lee H, Hou ST, Bazan NG, Haddad PS, Arnason JT, Bennett SA. Platelet activating factor-induced neuronal apoptosis is initiated independently of its G-protein coupled PAF receptor and is inhibited by the benzoate orsellinic acid. J. Neurochem., 103, 88–97 (2007).
- 29) Bazan NG. Lipid signaling in neural plasticity, brain repair, and neuroprotection. Mol. Neurobiol., 32, 89–103 (2005).
- 30) Tsuda M, Tozaki-Saitoh H, Inoue K. Platelet-activating factor and pain. Biol. Pharm. Bull., 34, 1159–1162 (2011).
- 31) Rink C, Khanna S. Significance of brain tissue oxygenation and the arachidonic acid cascade in stroke. Antioxid. Redox Signal., 14, 1889–1903 (2011).
- 32) Zhou Y, Huang J, He W, Fan W, Fang W, He G, Wu Q, Chu S, Li Y. N2 ameliorates neural injury during experimental ischemic stroke via the regulation of thromboxane A2 production. Pharmacol. Biochem. Behav., 124, 458–465 (2014).
- 33) Xu H, Wang E, Chen F, Xiao J, Wang M. Neuroprotective phytochemicals in experimental ischemic stroke: mechanisms and potential clinical applications. Oxid. Med. Cell. Longev., 2021, 6687386 (2021).
- 34) Wang X, Qin ZH, Shi H, Savitz SI, Qin AP, Jiang Y, Zhang HL. Protective effect of Ginkgolids (A + B) is associated with inhibition of NIK/IKK/IkappaB/NF-kappaB signaling pathway in a rat model of permanent focal cerebral ischemia. Brain Res., 1234, 8–15 (2008).
- 35) Desai A, Singh N, Raghubir R. Neuroprotective potential of the NF-kappaB inhibitor peptide IKK-NBD in cerebral ischemia-reperfusion injury. Neurochem. Int., 57, 876–883 (2010).
- 36) Jiang S, Li T, Ji T, Yi W, Yang Z, Wang S, Yang Y, Gu C. AMPK: potential therapeutic target for ischemic stroke. Theranostics, 8, 4535–4551 (2018).
- 37) Liang J, Luan Y, Lu B, Zhang H, Luo YN, Ge P. Protection of ischemic postconditioning against neuronal apoptosis induced by transient focal ischemia is associated with attenuation of NF-kappaB/p65 activation. PLOS ONE, 9, e96734 (2014).
- 38) Deng L, Wan H, Zhou H, Yu L, He Y. Protective effect of hydroxysafflor yellow A alone or in combination with acetylglutamine on cerebral ischemia reperfusion injury in rat: a PET study using (18)F-fuorodeoxyglucose. Eur. J. Pharmacol., 825, 119–132 (2018).
- 39) Shen Z, Zheng Y, Wu J, Chen Y, Wu X, Zhou Y, Yuan Y, Lu S, Jiang L, Qin Z, Chen Z, Hu W, Zhang X. PARK2-dependent mitophagy induced by acidic postconditioning protects against focal cerebral ischemia and extends the reperfusion window. Autophagy, 13, 473–485 (2017).
- 40) Cai Y, Yang E, Yao X, Zhang X, Wang Q, Wang Y, Liu J, Fan W, Yi K, Kang C, Wu J. FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia-reperfusion injury. Redox Biol., 38, 101792 (2021).
- 41) Nabavi SF, Sureda A, Sanches-Silva A, Pandima Devi K, Ahmed T, Shahid M, Sobarzo-Sanchez E, Dacrema M, Daglia M, Braidy N, Vacca RA, Berindan-Neagoe I, Gulei D, Barreca D, Banach M, Nabavi SM, Dehpour AR, Shirooie S. Novel therapeutic strategies for stroke: the role of autophagy. Crit. Rev. Clin. Lab. Sci., 56, 182–199 (2019).
- 42) Ni Y, Gu WW, Liu ZH, Zhu YM, Rong JG, Kent TA, Li M, Qiao SG, An JZ, Zhang HL. RIP1K contributes to neuronal and astrocytic cell death in ischemic stroke via activating autophagic-lysosomal pathway. Neuroscience, 371, 60–74 (2018).
- 43) Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, Kochanek PM, Jenkins LW, Ren J, Gibson G, Chu CT, Kagan VE, Clark RS. Starving neurons show sex difference in autophagy. J. Biol. Chem., 284, 2383–2396 (2009).
- 44) Li J, McCullough LD. Effects of AMP-activated protein kinase in cerebral ischemia. J. Cereb. Blood Flow Metab., 30, 480–492 (2010).
- 45) Guo FX, Wu Q, Li P, Zheng L, Ye S, Dai XY, Kang CM, Lu JB, Xu BM, Xu YJ, Xiao L, Lu ZF, Bai HL, Hu YW, Wang Q. The role of the LncRNA-FA2H-2-MLKL pathway in atherosclerosis by regulation of autophagy flux and inflammation through mTOR-dependent signaling. Cell Death Differ., 26, 1670–1687 (2019).
- 46) Chen S, Zhou L, Zhang Y, Leng Y, Pei XY, Lin H, Jones R, Orlowski RZ, Dai Y, Grant S. Targeting SQSTM1/p62 induces cargo loading failure and converts autophagy to apoptosis via NBK/Bik. Mol. Cell. Biol., 34, 3435–3449 (2014).
- 47) Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell, 137, 1001–1004 (2009).
- 48) Plaza-Zabala A, Sierra-Torre V, Sierra A. Autophagy and microglia: novel partners in neurodegeneration and aging. Int. J. Mol. Sci., 18, 598 (2017).
- 49) Raffo-Romero A, Arab T, Van Camp C, Lemaire Q, Wisztorski M, Franck J, Aboulouard S, Le Marrec-Croq F, Sautiere PE, Vizioli J, Salzet M, Lefebvre C. ALK4/5-dependent TGF-beta signaling contributes to the crosstalk between neurons and microglia following axonal lesion. Sci. Rep., 9, 6896 (2019).