Abstract
The oral bioavailability of berberine is quite low due to extensive first-pass metabolism. To increase the bioavailability of berberine (BBR), the efficacy of rectal administration that can avoid intestinal and hepatic first-pass metabolism partly was evaluated using BBR sulfate in rats. BBR sulfate was administered intravenously (1 mg/kg as BBR), orally (10 mg/kg as BBR) and rectally (1, 3, or 10 mg/kg as BBR) using Witepsol® H15 suppository base to evaluate bioavailability in rats. Concentrations of BBR in plasma were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS). When BBR sulfate was administered orally, the average oral bioavailability was 0.26%. When BBR sulfate was administered rectally, the average bioavailabilities were 17.0% at 1 mg/kg, 24.3% at 3 mg/kg, and 12.3% at 10 mg/kg as BBR, respectively. Thus, rectal administration of BBR sulfate greatly increased the bioavailability of BBR as compared with oral administration, which would also increase the pharmacological activities of BBR in vivo.
INTRODUCTION
Berberine (BBR) possesses a variety of pharmacological actions and is traditionally used for the treatment of a wide range of diseases, in addition to diarrhea and intestinal parasites.1,2) The clinical application of BBR, however, is limited due to its low oral bioavailability. The reported absolute oral bioavailability of BBR in rodents is less than 1%, and the urinary excretion rates of BBR are around 0.01–0.02% of the dose in rats and humans as reviewed.3,4) When BBR bioavailability in rats (0.356%) was analyzed pharmacokinetically, the low oral bioavailability of BBR was found to be due to the extensive intestinal first-pass metabolism (>98%).5) BBR is metabolized by plural cytochrome P450 enzymes (CYPs) including CYP1A2, CYP2D6, and CYP3A4.6)
In the present study, BBR sulfate was used as BBR and the rectal bioavailability of BBR sulfate was compared with the oral bioavailability in rats. BBR sulfate exhibits higher solubility than other BBR salts, and the rectal route can avoid both intestinal and hepatic first-pass metabolism at least partly.7–10)
MATERIALS AND METHODS
MaterialsBBR sulfate and Witepsol® H15, a suppository base, were obtained from Sigma-Aldrich Co., LLC (Tokyo, Japan) and Specialized Rx Products, LLC (MN, U.S.A.), respectively. Other reagents and solvents used were the highest quality available.
Animal StudiesMale Sprague–Dawley (SD) rats of 9 weeks old were fasted one night with free access to water before experiments. For intravenous administration, rats were anaesthetized with a combination anaesthetic (medetomidine 0.3 mg + midazolam 4.0 mg + butorphanol 5.0 mg/10 mL water/kg)11) and fixed on a waterbed kept at 37 °C. Rats were anaesthetized temporarily by inhalation of isoflurane vapour for blood sampling after oral and rectal administrations. The doses of BBR sulfate were 1 mg/mL/kg as BBR for intravenous injection. BBR sulfate (10 mg/mL/kg as BBR) was dissolved in 0.5% sodium carboxymethyl cellulose solution for oral administration. BBR sulfate was suspended in melted Witepsol® H15 at 42 °C and solidified at 5 °C. The suppository (rod-shaped with 5.0 mm diameter, 0.457 cm long for 100 g rats) was administered rectally to the site about 1 cm from the anus at a dose of 1, 3, or 10 mg as BBR/g suppository/kg, the anus was sealed with surgical glue and the animals were kept in a rat holder (KN-325-C-3, Natsume Seisakusho, Tokyo, Japan). The rat holder was positioned to keep the rat head 10 cm higher than its rectum to prevent the retrograde spread of the melted suppository. Blood sampling (0.1 mL each) was made from the jugular vein under light anaesthesia. The blood samples were centrifuged at 3000 × g at 4 °C for 10 min to obtain plasma samples. In the case of a 3 mg/kg BBR dose, the descending colon including the rectum (about 8 cm long from the anus) was isolated after 8-h blood sampling and killing rats with KCl-saturated solution. The colonic tissues isolated were finely chopped with surgical scissors and then homogenized using ULTRA-TURRAX® T-25 (IKA Japan Co., Ltd., Osaka, Japan) in a 5-fold weight of distilled water. Plasma (50 µL) and colonic homogenate (0.1 mL) samples were mixed with an equal volume of acetonitrile containing carbamazepine as an internal standard (IS) for deproteinization, the mixtures were centrifugated and the supernatants were subjected to the analysis of BBR concentrations by liquid chromatography-tandem mass spectrometry (LC-MS/MS).
Analysis of BBR by LC-MS/MSConcentrations of BBR in plasma were determined by LC-MS/MS (Model LCMS-8040, Shimadzu Corporation, Kyoto, Japan; Column: YMC-Triart C18, YMC Co., Ltd., Kyoto, Japan) using carbamazepine as IS. The mobile phase was a mixture of 0.1% formic acid and acetonitrile (50 : 50, v/v). The temperature of the sample cooler was 4 °C and the flow rate was isocratic (0.8 mL/min). The column was kept at 40 °C. Ionization was performed by positive electrospray mode at 250 °C and transitions from m/z = 336.10 to m/z = 292.10, and from m/z = 237.2 to m/z = 194.10 were recorded for BBR and carbamazepine (IS), respectively. The calibration curve for BBR was linear over the concentration range from 0.05 to 200 ng/mL.
Estimation of Pharmacokinetic ParametersPharmacokinetic parameters were determined as peak plasma concentration (Cmax), time to reach Cmax (Tmax), the area under the concentration–time curve from 0 to 8 h (AUC0–8h) for oral and rectal administrations, and AUC0–∞,iv from 0 to infinity after intravenous administration. Oral and rectal bioavailabilities of BBR sulfate were estimated by comparing AUC0–8h and AUC0–∞,iv normalized with BBR dose.
Statistical AnalysisData were expressed as the mean ± standard deviation (S.D.) of 3 or 4 trials for oral or rectal administrations, respectively. Differences in the mean values between groups were assessed using Student’s t-test or post hoc Tukey–Kramer test. p < 0.05 was considered statistically significant. Statical analysis was performed using the Statcel4 software (version 1.0, OMS Publishing Inc., Saitama, Japan).
Ethical ConsiderationsThis study was performed in accordance with the Guide for Animal Experimentation from the Committee of Research Facilities for Laboratory Animal Sciences, Hiroshima International University, which is in accordance with the Guidelines for Proper Conduct of Animal Experiments from the Science Council of Japan. The license number of this animal study was AE22-033 dated July 25, 2022.
RESULTS
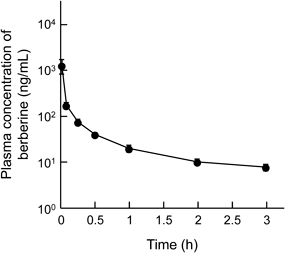
Intravenous Administration of BBRWhen BBR sulfate was administered intravenously at a dose of 1 mg/kg as BBR to rats, BBR disappeared from plasma according to the two-compartment model (Fig. 1). Some pharmacokinetic parameters such as Cmax, AUC0–3h, and AUC0–∞ are listed in Table 1. The value of AUC0–∞,iv of plasma BBR was used to estimate the bioavailabilities of BBR after oral and rectal administrations.
Table 1. Peak Plasma Concentrations (
Cmax), Time to Reach
Cmax (
Tmax) and
AUC Values, and Oral and Rectal Bioavailabilities of BBR Sulfate after Oral and Rectal Administrations in Rats
| Parameters/Dose as BBR | i.v. | p.o. | Rectal |
|---|
| (1 mg/kg) | (10 mg/kg) | (1 mg/kg) | (3 mg/kg) | (10 mg/kg) |
|---|
| Cmax (ng/mL) | 2490 ± 1054 | 0.94 ± 0.10 | 9.62 ± 5.04 | 29.4 ± 7.2 | 57.0 ± 18.0 |
| Tmax (h) | — | 2.0 ± 0.8 | 2.0 ± 2.3 | 1.5 ± 1.5 | 0.6 ± 0.2 |
| AUC0–t (ng·h/mL) | 147.6 ± 37.6 | 4.1 ± 0.2 | 27.3 ± 7.9 | 117.1 ± 26.8 | 197.3 ± 71.3 |
| AUC0–∞ (ng·h/mL) | 160.7 ± 35.9 | — | — | — | — |
| Bioavailability (%) | 100 | 0.26 ± 0.01 | 17.0 ± 4.9** | 24.3 ± 5.6** | 12.3 ± 4.4*,† |
Each value represents the mean ± S.D. of 3 or 4 trials for oral or rectal administrations, respectively. In estimating the oral and rectal bioavailabilities of berberine (BBR), average values of AUC0–∞ after intravenous (i.v.) administration and value of AUC0–8h after oral (p.o.) and rectal administrations of BBR sulfate were used. * p < 0.05 and ** p < 0.01: significantly different from p.o. of BBR. †p < 0.05: significantly different from rectal administration (3 mg/kg) of BBR.
BBR sulfate was administered orally at a dose of 10 mg/kg as BBR to rats. The Cmax of BBR in plasma was observed in a range from 1 to 3 h after administration. The average oral bioavailability of BBR sulfate estimated was 0.26 ± 0.01% in rats (Fig. 2, Table 1).
Rectal Administration of BBRBBR sulfate was administered rectally at doses of 1, 3, and 10 mg/kg as BBR using Witepsol® H15 as a suppository base. The plasma concentrations of BBR after rectal administration at a dose of 10 mg/kg were far higher than those after oral administration (Fig. 2). The value of Cmax of plasma BBR increased with an increase in the dose of BBR sulfate, and the average value of Tmax after rectal administration became shorter from 2.0 to 0.6 h when the dose of BBR sulfate increased. The amounts of BBR that remained in the colonic lumen 8 h after rectal administration (dose, 3 mg/kg) were 9.73 ± 4.28% (n = 4) of the dose. The average rectal bioavailabilities of BBR estimated were 17.0% at doses of 1 mg/kg, 24.3% at 3 mg/kg, and 12.3% at 10 mg/kg as BBR, respectively, which corresponded to 47.3–93.5 fold of oral administration in rats (Table 1).
DISCUSSION
The absolute oral bioavailabilities (F) of raw BBR in rats reported were all less than 1% of the dose, and in the present study, the estimated oral bioavailability of BBR sulfate was 0.27%, in good agreement with previously reported data.3–5) To increase the oral bioavailability of BBR, a substrate for P-glycoprotein (P-gp) and plural CYPs, a variety of dosage formulations of BBR has been developed by overcoming absorption-reducing factors of BBR such as extensive intestinal first-pass metabolism and insufficient intestinal absorption due to the low solubility and P-gp-mediated efflux transport.3–6,12) Among them, formulations containing surfactants exhibiting P-gp and CYPs inhibition and formulations containing an absorption enhancer that facilitates paracellular transport and avoids intestinal first-pass metabolism increased bioavailability by more than 10-fold of BBR alone.4) In particular, the latter formulations containing absorption enhancers such as sodium caprate (C10) and sodium deoxycholate (NaDC) showed greater oral bioavailability of BBR (around 40-fold of BBR alone).13)
In the present study, to avoid or reduce intestinal first-pass metabolism of BBR, the efficacy of rectal administration of BBR was examined in rats. The vascular system of the rectum is as follows: rectal drainage is controlled by three veins, namely the superior, middle, and inferior rectal veins. The superior rectal vein drains the upper part of the rectum (via the inferior mesenteric vein) into the portal venous system, and the middle and inferior rectal vein drains the lower part of the rectum into the internal iliac vein (via the internal pudendal vein) for systemic circulation.10) It was reported that rectal administration of nitroglycerin gave a bioavailability of 26.7 ± 7.0% compared to 1.8 ± 0.9% from oral dosing. When the rectal exposure length to nitroglycerin was restricted to 3.5 cm from the anus, the bioavailability was 83.5%, and when the rectal exposure length was restricted to 2.0 cm from the anus, the bioavailability was 91.2% in rats.8) Similar results were also reported with lidocaine in rats.9) In the present study, the rat holder was set to keep the rat head 10 cm higher than the bottom portion to prevent the retrograde spread of the melted suppository. As BBR, BBR sulfate was used because BBR sulfate exhibits higher solubility (277.0 mg/mL at 37 °C), than other BBR salts such as BBR chloride (2.64 mg/mL in water at 37 °C) and the effect of NaCl (salting-out) on the solubility is small.7) Rectal administration of BBR sulfate greatly increased BBR bioavailability as compared with that after oral bioavailability (Table 1), possibly due to the avoidance of first-pass metabolism at least partly. In addition, the higher absorption rate (Fa) of BBR sulfate after rectal administration than that after oral administration may also be involved. The reported Fa value of BBR after oral administration (100 mg/kg dose) was 44% in rats.5) In the present study, 9.73% of BBR dosed was recovered in the descending colon 8 h after rectal administration (3 mg/kg), which may suggest a higher Fa value of BBR after rectal administration than that after oral administration. A further study employing the in-situ rectal loop is required to estimate the precious Fa value of BBR in the rectum and the relationship between the exposure area of BBR in the rectum and the extent of first-pass metabolism as reported previously using nitroglycerine and lidocaine.8,9) The rectum is a closed space with constant neutral pH compared to the gastrointestinal lumen, and therefore, excipients can act effectively by maintaining high concentrations. Various acidic compounds including NaDC and C10 are known to act as absorption enhancers in the rectum and they may further increase the rectal absorption of BBR sulfate than oral administration of BBR.13–15)
In conclusion, rectal administration that can reduce first-pass metabolism can increase the bioavailability and possibly pharmacological activities of BBR greatly as compared with oral administration.
Conflict of Interest
The authors declare no conflict of interest.
REFERENCES
- 1) Neag MA, Mocan A, Echeverría J, Pop RM, Bocsan CI, Crişan G, Buzoianu AD. Berberine: botanical occurrence, traditional uses, extraction methods, and relevance in cardiovascular, metabolic, hepatic, and renal disorders. Front. Pharmacol., 9, 557 (2018).
- 2) Song D, Hao J, Fan D. Biological properties and clinical applications of berberine. Front. Med., 14, 564–582 (2020).
- 3) Murakami T, Bodor E, Bodor N. Approaching strategy to increase the oral bioavailability of berberine, a quaternary ammonium isoquinoline alkaloid: part 1. Physicochemical and pharmacokinetic properties. Expert Opin. Drug Metab. Toxicol., 19, 129–137 (2023).
- 4) Murakami T, Bodor E, Bodor N. Approaching strategy to increase the oral bioavailability of berberine, a quaternary ammonium isoquinoline alkaloid: part 2. Development of oral dosage formulations. Expert Opin. Drug Metab. Toxicol., 19, 139–148 (2023).
- 5) Liu YT, Hao HP, Xie HG, Lai L, Wang Q, Liu CX, Wang GJ. Extensive intestinal first-pass elimination and predominant hepatic distribution of berberine explain its low plasma levels in rats. Drug Metab. Dispos., 38, 1779–1784 (2010).
- 6) Li Y, Ren G, Wang YX, Kong WJ, Yang P, Wang YM, Li YH, Yi H, Li ZR, Song DQ, Jiang JD. Bioactivities of berberine metabolites after transformation through CYP450 isoenzymes. J. Transl. Med., 9, 62 (2011).
- 7) Miyazaki S, Oshiba M, Nadai T. Dissolution properties of salt forms of berberine. Chem. Pharm. Bull., 29, 883–886 (1981).
- 8) Kamiya A, Ogata H, Fung HL. Rectal absorption of nitroglycerin in the rat: avoidance of first-pass metabolism as a function of rectal length exposure. J. Pharm. Sci., 71, 621–624 (1982).
- 9) de Leede LG, de Boer AG, Roozen CP, Breimer DD. Avoidance of “first-pass” elimination of rectally administered lidocaine in relation to the site of absorption in rats. J. Pharmacol. Exp. Ther., 225, 181–185 (1983).
- 10) Rathi R, Sanshita, Kumar A, Vishvakarma V, Huanbutta K, Singh I, Sangnim T. Advancements in rectal drug delivery systems: clinical trials, and patents perspective. Pharmaceutics, 14, 2210 (2022).
- 11) Tashiro M, Tohei A. Recommended doses of medetomidine-midazolam-butorphanol with atipamezole for preventing hypothermia in mice. J. Vet. Med. Sci., 84, 445–453 (2022).
- 12) Kwon M, Lim DY, Lee CH, Jeon JH, Choi MK, Song IS. Enhanced intestinal absorption and pharmacokinetic modulation of berberine and its metabolites through the inhibition of P-glycoprotein and intestinal metabolism in rats using a berberine mixed micelle formulation. Pharmaceutics, 12, 882 (2020).
- 13) Fan D, Wu X, Dong W, Sun W, Li J, Tang X. Enhancement by sodium caprate and sodium deoxycholate of the gastrointestinal absorption of berberine chloride in rats. Drug Dev. Ind. Pharm., 39, 1447–1456 (2013).
- 14) Murakami T, Sasaki Y, Yamajo R, Yata N. Effect of bile salts on the rectal absorption of sodium ampicillin in rats. Chem. Pharm. Bull., 32, 1948–1955 (1984).
- 15) Nishimura K, Nozaki Y, Yoshimi A, Nakamura S, Kitagawa M, Kakeya N, Kitao K. Studies on the promoting effects of carboxylic acid derivatives on the rectal absorption of beta-lactam antibiotics in rats. Chem. Pharm. Bull., 33, 282–291 (1985).