Abstract
Liver cancer is one of the most aggressive tumors and one of the most common malignant tumors which seriously threatens human health. Traditional Chinese medicine (TCM) was reported to resist the proliferation and metastasis of liver cancer cells. In this study, we aimed to explore the potential anti-cancer effect of Polygonatum sibiricum polysaccharide (PSP) on the tumor immune microenvironment in liver cancer cells. HepG2 and Hep3B cells were pretreated in the absence or the presence of PSP (20, 50, 100 µg/mL) for a period of 24 h. Subsequently, dendritic cells (DCs) were co-cultured with HepG2 and Hep3B cell supernatant to investigate the effect of PSP on the tumor microenvironment. The results showed that PSP dose-dependently inhibited proliferation and promoted apoptosis of HepG2 and Hep3B cells. Meanwhile, PSP dose-dependently inhibited migration, invasion, and epithelial-to-mesenchymal transition (EMT) of liver cancer cells. In addition, PSP dose-dependently induced inflammatory response of DCs, characterized by increases of interleukin (IL)-6, IL-1β, and tumor necrosis factor (TNF)-α in DCs. Mechanically, PSP dose-dependently reduced the activation of the Toll-like receptor 4 (TLR4)/Signal transducer and activator of transcription 3 (STAT3) and noncanonical nuclear factor-kappa B (NF-κB) signaling pathways. TLR4 agonist lipopolysaccharide (LPS) reversed the anti-oncogenic effects of PSP in liver cancer cells. Taken together, PSP inhibited liver cancer in a simulated tumor microenvironment by eliminating TLR4/STAT3 pathway. PSP promises an important and useful alternative to liver cancer treatment.
INTRODUCTION
Primary liver cancer was a kind of common cancer around the world.1) Multiple factors and procedures were participating in the hepatocarcinogenesis. Up to now, research suggested that hepatocarcinogenesis was tightly linked to liver diseases, such as chronic virus hepatitis (hepatitis B virus (HBV) and hepatitis C virus (HCV)), alcoholic hepatitis or fibrosis, and non-alcoholic fatty liver diseases.2,3) In general, long-term chronic hepatic injury, injury-induced chronic liver inflammation, and compensatory hepatocyte proliferation play critical roles in hepatocarcinogenesis.4,5) There were plenty of molecules and pathways involved in hepatocarcinogenesis, which made the underlying mechanisms extremely complex.6) Although liver cancer could be surgically resected, drug treatment had an irreplaceable function for patients in advanced stages.7,8) However, chemotherapy agents were still limited by many disadvantageous factors, such as side effects and drug resistance. It was urgent to find more effective therapeutic regimens.
Traditional Chinese medicine (TCM) was believed to have lower side effects and treat disease by the comprehensive effect of its components. TCM had advantages in the treatment of complex diseases and chronic diseases, including liver cancer.9,10) Polygonatum was a perennial herb of the genus Polygonatum Mill in the Liliaceae family which contains polysaccharides, saponins, flavonoids, lignans amino acid quinone compounds, vitamins, alkaloids, and a variety of trace elements.11,12) Polygonatum had anti-oxidation, anti-tumor hypoglycemic, immune regulation, anti-bacterial anti-inflammatory, anti-viral, and other functions, which was a plant with high medicinal value and nutritional value. Polygonatum sibiricum polysaccharide (PSP) is the main active ingredient in Polygonatum, it is light yellow solid powder, slightly soluble in cold water, easily soluble in hot water.12) It was found that the sweet taste of polygonatum was attributed to PSP, which made the food more appealing.13,14) PSP chemical composition analysis confirmed that PSP were composed of a branched homogalactan and branched galactomannans.11) PSP has anti-oxidation, anti-tumor hypoglycemic, immune regulation, anti-inflammatory, and anti-viral effects.15,16) It was found that PSP pretreatment protects against N-methyl-4-phenyl pyridine (MPP+)-induced neurotoxicity through the protein kinase B (Akt)/mammalian target of rapamycin (mTOR) and nuclear factor-E2-related factor 2 (Nrf2) pathways.17) PSP also had a powerful protective effect on gentamicin (GM)-induced acute kidney injury in rats by decreasing the expression of NGAL or KIM-1.18) Interestingly, a previous study proved that PSP plays an anti-cancer effect via the Toll-like receptor 4 (TLR4)/mitogen-activated protein kinase (MAPK)/nuclear factor-kappa B (NF-κB) signaling pathways.15) However, whether PSP plays a direct role in the treatment of liver cancer remains ambiguous. In this study, we explored the effect and underlying mechanisms of PSP on the inflammatory tumor microenvironment and liver cancer development in vitro.
MATERIALS AND METHODS
Cell CultureWe followed the methods of Bu X.19) Human liver cancer cell lines, HepG2 (CL-0103) and Hep3B (CL-0102) were obtained from ProCell (Wuhan, China). HepG2 cells were authenticated by STR profiling. The cells were cultured (37 °C, 5% CO2) in Modified Eagle’s Medium (MEM) medium (Gibco, Carlsbad, CA, U.S.A.) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone, South Logan, UT, U.S.A.) and 1% P/S.
Cell TreatmentHepG2 and Hep3B cells were seeded into 96-well multi plates (2 × 105 cells/well). cells were pretreated in the absence or the presence of PSP (Dekang Biotechnology, Ningbo, China; 20, 50, 100 µg/mL) for a period of 24 h. The concentration gradient settings of PSP refer to the previous article20) with some modifications. For some experiments, HepG2 and Hep3B cells were incubated with 1 µg/mL TLR4 agonist lipopolysaccharide (LPS; InvivoGen, San Diego, CA, U.S.A.) for 24 h.
The Preparation of Tumor-Cell SupernatantHepG2 and Hep3B cells were treated with PSP and/or LPS for 24 h. Then, HepG2 and Hep3B cell supernatant was prepared by centrifugation (4000 × g, 4 °C) for 10 min. To activate human dendritic cells (DCs; CP-H187B, ProCell, Wuhan, China), cells were exposed to the RPMI-1640 medium supplemented with 10% heat-inactivated FBS (Hyclone, South Logan, UT, U.S.A.) and 1% P/S, and were passaged 2–3 times.
DCs in the logarithmic growth phase were collected, cells were digested with trypsin, and the cell concentration was adjusted to 2.5 × 104/mL. DC cells were seeded in 96-well plates with a volume of 200 µL per well, and the supernatant of HepG2 and Hep3B cells was added from day 0 of cell culture respectively.
Cell Proliferation AssayAccording to the manufacturer’s instructions, cell proliferation of HepG2 and Hep3B cells was measured by the cell counting kit-8 (CCK-8, Dojindo, Japan) assay. In short, a total of 2.0 × 103 cells/well were seeded into 96-well plates, with 10 µL of CCK-8 solution added to each well containing 10 µL of MEM medium and incubated at 37 °C under 5% CO2 for 3 h. After 48 h incubation, absorbance at 450 nm was recorded by a microplate reader (Spectra Max Plus 384, Molecular Devices, Sunnyvale, CA, U.S.A.).
Wound Healing AssayHepG2 and Hep3B cells were seeded into 6-well plates for 24 h to reach 80–90% confluence. The well was scratched with a 10 µL pipette to create a single wound in the center of the cell monolayer. Subsequently, cells were cultured in a serum-free MEM medium for 24 h at 37 °C. The wound healing distance was measured under an Olympus X51 inverted microscope (Olympus, Tokyo, Japan). Relative wound recovery (%) = (initial wound width-wound width at 24 h)/initial wound width × 100.
Transwell AssayTranswell filters (BD Biosciences, San Jose, CA, U.S.A.) were filled with the 4 °C pre-chilled serum-free medium to form the chamber. HT29 and SW480 cells were suspended in serum-free medium (3.0 × 105/mL) and seeded into the upper chamber. A culture medium containing 10% FBS was added to the basolateral chamber and left to incubate at 37 °C for 24 h. Then, transwell cultures were washed twice with sterile phosphate buffered saline (PBS), fixed with methanol for 30 min, and then dyed with 0.1% crystal violet for 30 min at room temperature. Cell invasion was observed under an Olympus X51 inverted microscope (Olympus).
Flow Cytometry AssayThe apoptosis of HepG2 and Hep3B cells was analyzed using an Annexin V- according to the manufacturer’s protocol. Briefly, the cells were washed with PBS (Invitrogen, Carlsbad, CA, U.S.A.), and adjusted the cell concentration to 1.0 × 106 cells/mL. The cells were subsequently suspended in a 150 µL buffering solution. Subsequently, the cells were stained with 10 µg/mL Annexin V-fluorescein isothiocyanate (FITC) and 5 µL propidium iodide (PI) at 4 °C for 20 min in darkness. Apoptotic cells were then analyzed by a BD FACSCelesta™ Flow Cytometer (Becton, Dickinson, and Company, NJ, U.S.A.).
RNA Extraction and RT Quantitative PCR (RT-qPCR)TRIzol (Invitrogen) reagent was used to extract total RNA from CDs according to the manufacturer’s protocol. RT-qPCR was performed by using the SYBR Premix Ex TaqTMII kit (TaKaRa, Tokyo, Japan). RT-qPCR was performed with a program of 5 min at 95 °C and then 45 cycles at 95 °C (30 s), 95 °C (5 s), 55 °C (30 s), and 72 °C (30 s). The cycle threshold (CT) values of the samples were analyzed using Thermo Scientific PikoReal software 2.1 (Thermo Fisher Scientific, Waltham, MA, U.S.A.). The 2 −ΔΔCT method (ΔΔCT = ΔCTtreatment − ΔCTcontrol and ΔCT = Cttarget − Ctreference.) was used to calculate the relative expression. Each candidate gene was internally normalized against β-actin. The primer sequences used are shown below: interleukin (IL)-6: forward, 5′-AAT GAG GAG ACT TGC CTG GT-3′ reverse, 5′-GCA GGA ACT GGA TCA GGA CT-3′. IL-1β: forward, 5′-CAC CTT CTT TTC CTT CAT CTT TG-3′ reverse, 5′-GTC GTT GCT TGT CTC TCC TTGTA-3′. Tumor necrosis factor (TNF)-α: forward, 5′-TCT TCT CGA ACC CCG AGT GA-3′ reverse, 5′-TAT CTC TCA GCT CCA CGC CA-3′. β-Actin: forward, 5′-CAT GTA CGT TGC TAT CCA GGC-3′, reverse 5′-CTC CTT AAT GTC ACG CAC GAT-3′.
Western Blot AnalysisHepG2 and Hep3B cells were lysed in radio immunoprecipitation assay (RIPA) buffer (Signaling Technology, Inc., Danvers, MA, U.S.A.) to collect total proteins. The protein content was quantitated using a bicinchoninic acid (BCA) kit (Beyotime, Shanghai, China). Total protein (30 µg/sample) was separated via 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and transferred to nitrocellulose membranes (Pall Life Sciences, New York, U.S.A.). The membranes were blocked with 5% nonfat dry milk for 1 h to prevent nonspecific binding of antibodies. After being probed with primary antibodies overnight at 4 °C, the membranes were washed and incubated with appropriate peroxidase-conjugated secondary antibodies. The corresponding protein primary antibodies were as follows: Bcl-2 (No. ab32124, Abcam, Cambridge, MA, U.S.A.; 1 : 1000), Bax (No. ab32503, Abcam; 1 : 1000), E-cadherin (No. A20798, Abclonal, Wuhan, China; 1 : 500), N-cadherin (No. A0433, Abclonal; 1 : 500), vimentin (No. A19607, Abclonal; 1 : 500), TLR4 (No. A15258, Abclonal; 1 : 500), STAT3 (No. A1192, Abclonal; 1 : 500), p-STAT3 (No.AP0070, Abclonal; 1 : 500), p65 (No. ab32536, Abcam; 1 : 1000), p-p65 (No. ab76302, Abcam; 1 : 1000), and β-actin (No. ab8227, Abcam; 1 : 1000). Finally, the bands were visualized using the ECL system (Thermo Scientific, Rockford, IL, U.S.A.).
Immunofluorescence (IF) StainingHepG2 and Hep3B cells were fixed with 4% paraformaldehyde for 20 min at room temperature and subsequently treated with 0.25% TritonX-100 for 15 min. After blocking with 5% bovine serum albumin (BSA) at 37 °C for 1 h, the cells were incubated with anti-NF-кB p65 (BS-0465R; Bioss, Beijing, China; 1: 100) antibody overnight at 4 °C. The cells were incubated with FITC-conjugated goat anti-rabbit immunoglobulin G (IgG) (GB22303; Servicebio, Wuhan, China; 1 : 100) at 37 °C for 1 h and then stained with 4′-6-diamidino-2-phenylindole (DAPI) for 5 min. The cell staining was observed under a fluorescence microscope after sealing the slides with a fluorescence microscope (Olympus OlyVIA, Tokyo, Japan).
Statistical AnalysisAll cell experiments were repeated at least in triplicate. The data were represented as means ± standard deviation (S.D.). Statistical analysis was performed with the SPSS software (version 19.0, SPSS Inc., Chicago, IL, U.S.A.). One-way ANOVA with Tukey’s post hoc test was used for statistical analysis. Differences with a p < 0.05 were considered to indicate statistically significant.
RESULTS
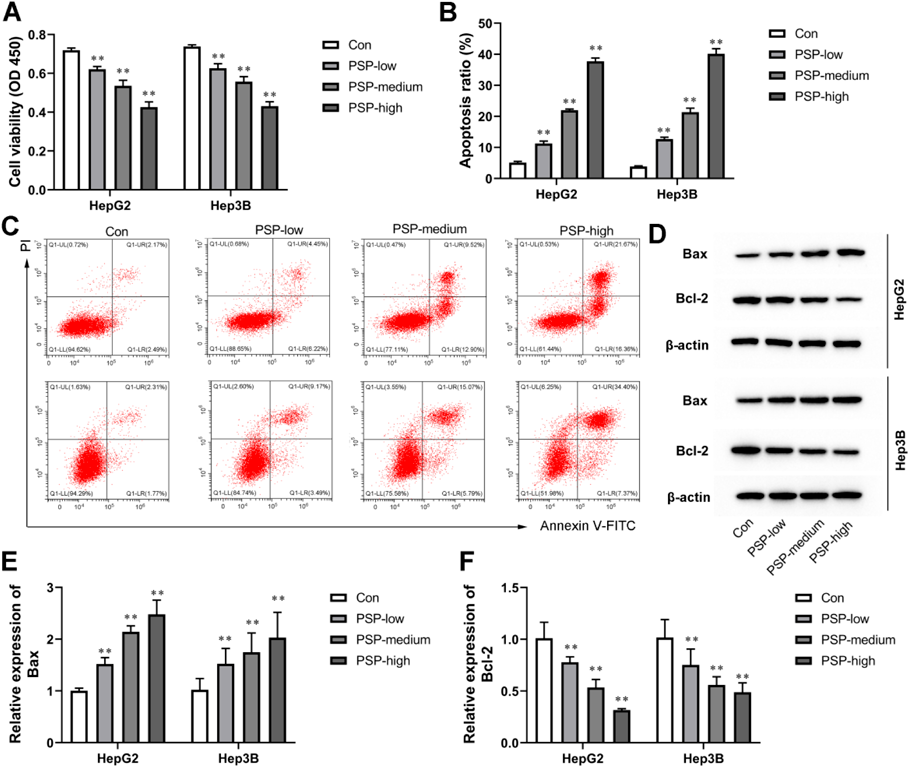
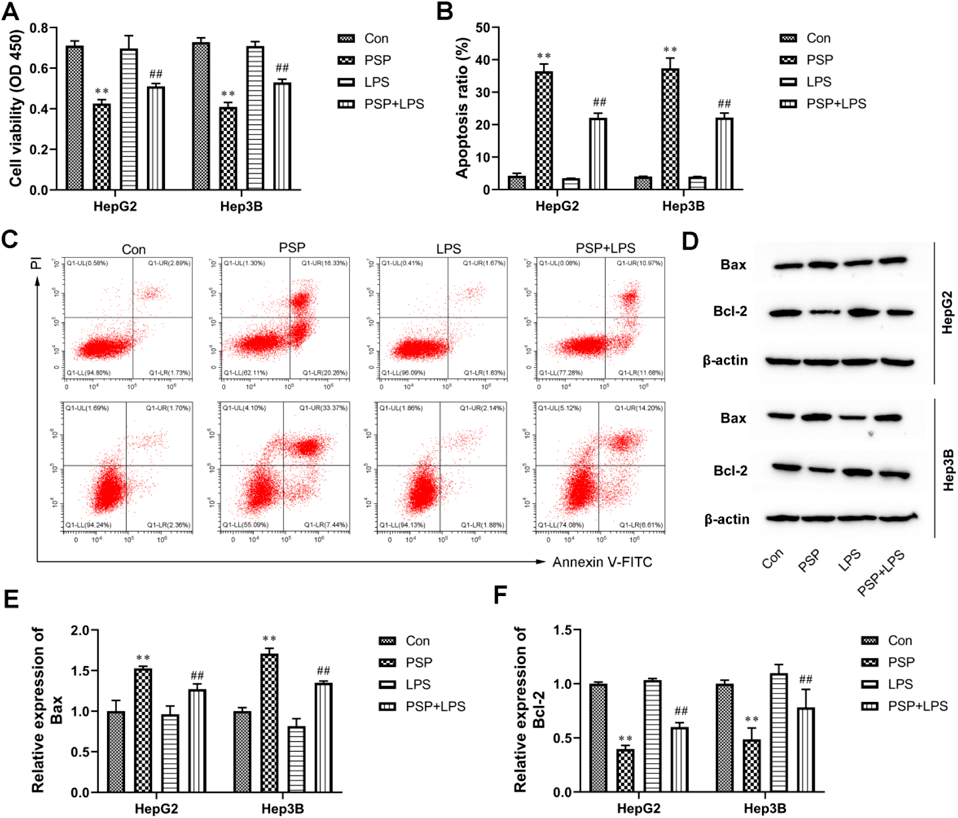
PSP Dose-Dependently Inhibited Proliferation and Promoted Apoptosis of Liver Cancer CellsTo explore the function of PSP in liver cancer, different concentrations of PSP (20, 50, and 100 µg/mL) were used to treat HepG2 and Hep3B cells for 24 h. The effect of PSP on cell proliferation was evaluated by a CCK-8 assay. Our results indicated that PSP dose-dependently inhibited cell proliferation of HepG2 and Hep3B cells compared with the control group (Fig. 1A). In addition, PSP significantly promoted cell apoptosis of HepG2 and Hep3B cells (Figs. 1B, C). To confirm whether PSP treatmentregulates the expression of apoptotic-related proteins, the protein expression of Bax and Bcl-2 was determined. Compared with the control group, the expression of Bax was increased, whereas the expression of Bcl-2 was decreased in PSP-treated HepG2 and Hep3B cells (Figs. 1D–F). Collectively, these results demonstrate that PSP dose-dependently inhibited liver cancer cell growth.
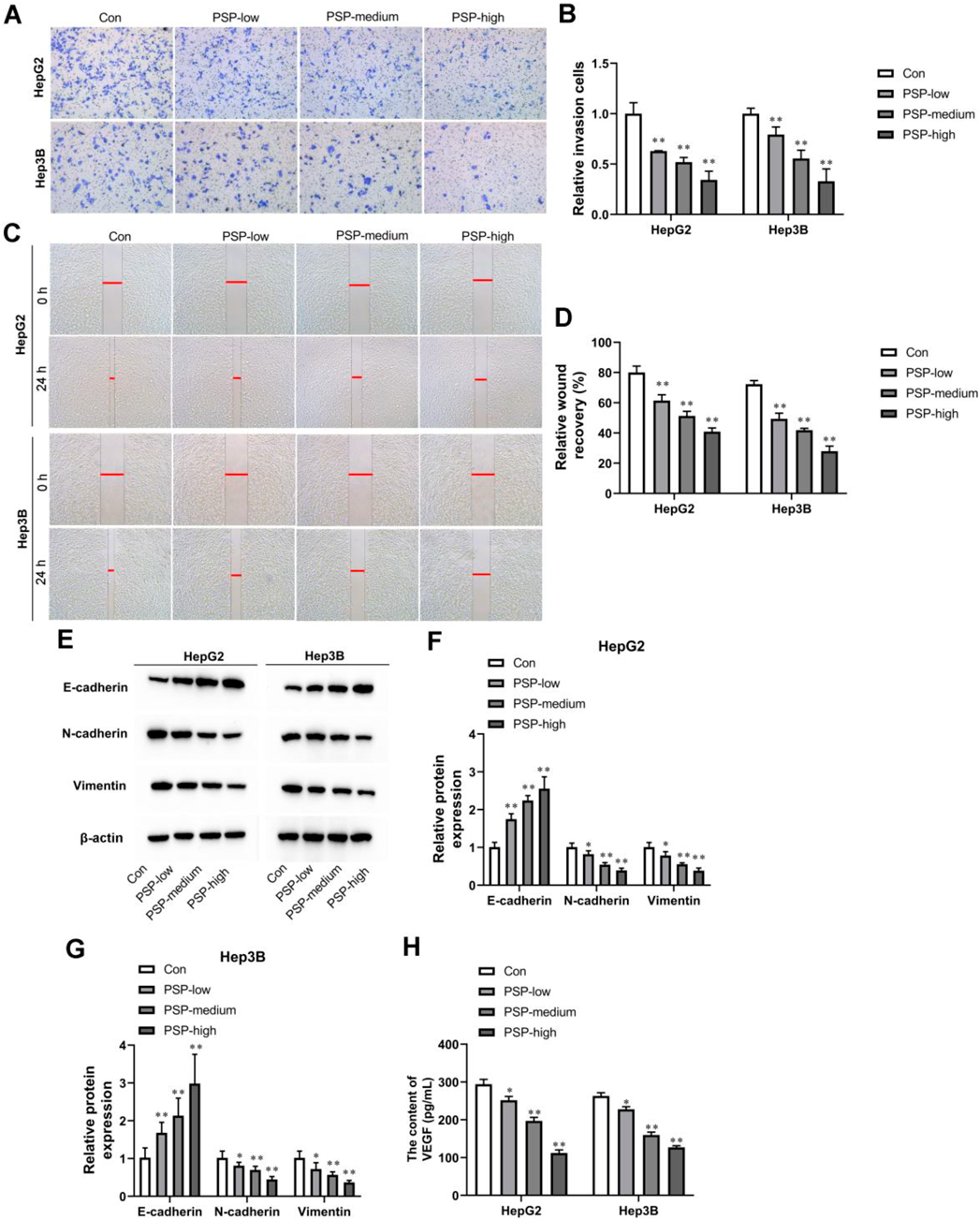
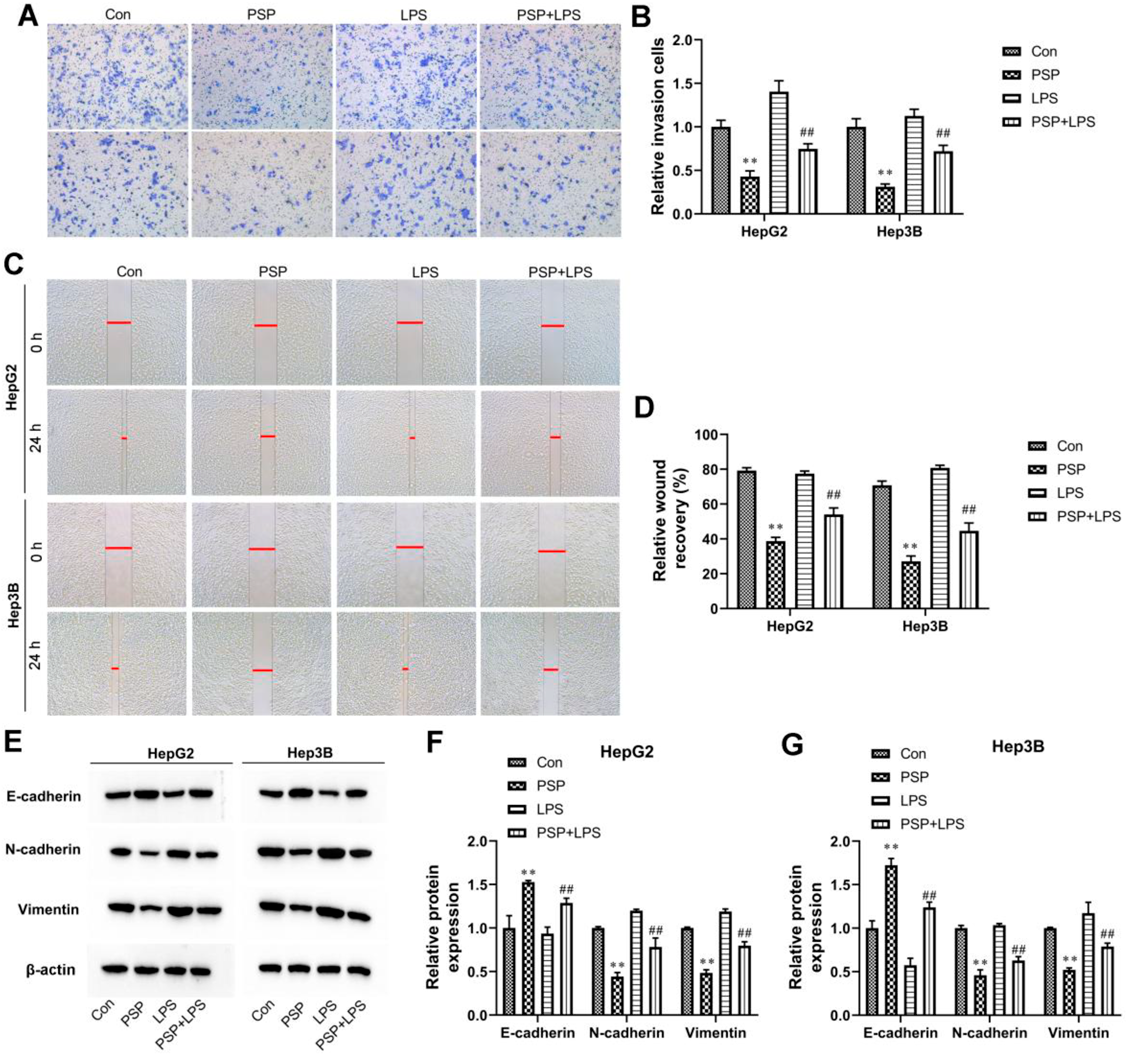
PSP Dose-Dependently Inhibited Migration, Invasion, and Epithelial-to-Mesenchymal Transition (EMT) of Liver Cancer CellsTranswell assay results showed that invasion cells were noticeably declined in HepG2 and Hep3B cells treated with PSP (Figs. 2A, B). Meanwhile, in wound healing assay, PSP dose-dependently suppressed cell migration of HepG2 and Hep3B cells (Figs. 2C, D). In addition, compared with the control group, the expression of epithelial marker E-cadherin was dose-dependently increased, whereas the expression of one epithelial marker N-cadherin and mesenchymal markers vimentin was dose-dependently decreased in PSP-treated HepG2 and Hep3B cells (Figs. 2E, G). We concluded that PSP dose-dependently inhibited cell migration, invasion, and EMT in HepG2 and Hep3B cells. In addition, we examined vascular endothelial growth factor (VEGF) expression in the supernatants of HepG2 and Hep3B cells. The results showed that PSP decreased VEGF production in a dose-dependent manner (Fig. 2H).
PSP Dose-Dependently Induced Inflammatory Response of DCsTo further investigate the effect of PSP on the tumor microenvironment, we co-cultured DCs with PSP-treated HepG2 or Hep3B cells. It is known that DCs plays an important role for cancer cells in antigen presentation. Figure 3A shows the expression of DC maturation markers CD80, CD83, and human leukocyte antigen (HLA)-DR. Flow cytometry suggested that both in DCs co-cultured with HepG2 or Hep3B cell supernatant, PSP dose-dependently increased the expression CD80, CD83, and HLA-DR (Fig. 3A). Moreover, the mRNA levels of IL-6, IL-1β, and TNF-α in DCs were enhanced by 20, 50, and 100 µg/mL PSP (Figs. 3B, C). Additionally, Western blot analysis showed that the increases in p-p65 protein levels were observed in DCs co-cultured with HepG2 or Hep3B cell supernatant (Figs. 3D–F). Taken together, these results strongly suggest that PSP could dose-dependently stimulate DC maturation and activate the inflammatory response of DCs co-cultured with HepG2 or Hep3B cell supernatant.
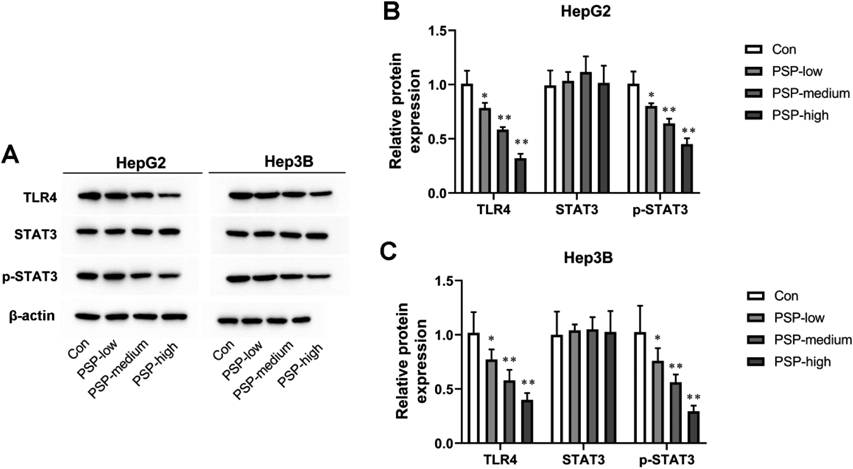
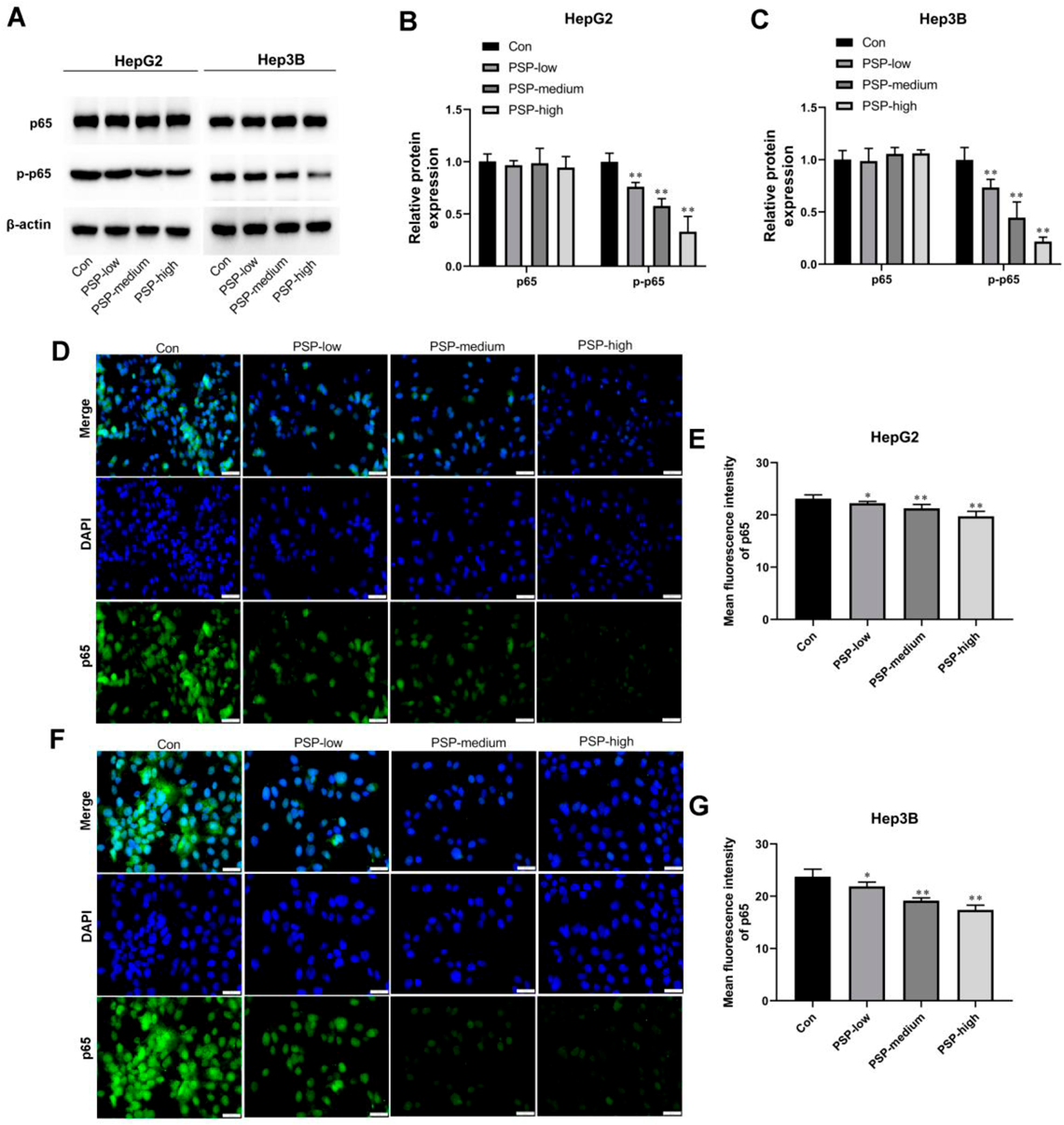
PSP Dose-Dependently Reduced the Activation of the TLR4/STAT3 and NF-κB Signaling PathwaysTLR4/STAT3 signaling pathway played an important role in liver cancer progression. Next, we wondered whether PSP could activate the TLR4/STAT3 signaling pathway in HepG2 and Hep3B cells. The increases in TLR4 and p-STAT3 protein levels were observed in PSP-treated HepG2 and Hep3B cells compared with the control group (Figs. 4A–C). These results strongly suggest that PSP could suppressthe TLR4/STAT3 signaling pathway in HepG2 and Hep3B cells (Figs. 4A–C). In addition, we investigated the effects of PSP on the activation of NF-κB. As shown in Figs. 5A–C, PSP increased the phosphorylation of p65 in HepG2 and Hep3B cells in a dose-dependent manner. IF results showed that PSP promoted the nuclear translocation of p65 in HepG2 and Hep3B cells (Figs. 5D–G).
TLR4 Agonist LPS Reversed the Anti-oncogenic Effects of PSP in Liver Cancer CellsTo investigate whether TLR4/STAT3 signaling pathway mediates the anti-oncogenic effects of PSP, the HepG2 and Hep3B cells were co-treated with 100 µg/mL PSP and 1 µg/mL TLR4 agonist LPS. As shown in Fig. 6A, the CCK-8 assay demonstrated that PSP inhibited the proliferation of HepG2 and Hep3B cells, which was reversed by LPS treatment. Meanwhile, LPS weakened cell apoptosis of HepG2 and Hep3B cells compared with the PSP-treated groups (Figs. 6B, C). Moreover, PSP-treated HepG2 and Hep3B cells displayed an increase in Bax expression and a decrease in Bcl-2 expression, which were both reversed by LPS (Figs. 6D, E). In addition, the transwell assay showed that PSP inhibited HepG2 and Hep3B cell invasion, which was significantly strengthened by LPS treatment (Figs. 7A, B). Meanwhile, the results of the wound healing assay manifested that the healing of the scratch was significantly induced following LPS co-treatment compared with the PSP-treated groups (Figs. 7C, D). In addition, the expression of EMT-related proteins N-cadherin and Vimentin were eliminated by PSP treatment, which was significantly strengthened by LPS treatment (Figs. 7E–G). In contrast, the E-cadherin expression was opposite in HepG2 and Hep3B cells (Figs. 7E–G). Taken together, our results indicated that activation of the TLR4/STAT3 signaling pathway reversed the effects caused by PSP treatment in liver cancer cells.
DISCUSSION
In this study, PSP dose-dependently inhibited proliferation, migration, invasion, and EMT and promoted apoptosis of liver cancer cells. Meanwhile, PSP increased the expression of CD80, CD83, and HLA-DR in DCs in vitro. PSP dose-dependently reduced the activation of the TLR4/STAT3 signaling pathway. Moreover, TLR4 agonist LPS reversed the anti-oncogenic effects of PSP in liver cancer cells. These findings demonstrated that the mechanism underlying the improving effect of PSP on tumor development and the immunosuppression of tumor microenvironment in liver cancer is related to the TLR4/STAT3 pathway.
As a hot spot for research recurrently, the tumor microenvironment played an important role in the recurrence and metastasis of liver cancer.21) The tumor microenvironment is the microenvironment for carcinogenesis and progression which consisted of tumor cells, fibroblasts, endothelial cells innate and acquired immune cells structure cells of tumor vessels, tissue-specific mesenchymal, and their metabolites products.22) Some studies showed that immunological imbalance has a close relationship with the potential invasion of tumor cells and the prognosis of the treatment.23,24) A previous study suggested that DCs as professional antigen-presenting cells were related to liver cancer development.25) Interestingly, PSP has been proven to be able to promote immune organ cells to enter into S and G2/M phases as well as inhibited the apoptosis in the spleen, thymus, and bursa of Fabricius.26) Cetyltrimethylammonium bromide-modified Polygonatum sibiricum polysaccharide cubosomes (CTAB-modified PSP-Cubs/OVA) could promote the secretion of related cytokines and significantly activate dendritic cells.27) In our results, PSP dose-dependently stimulated DC maturation to activate the inflammatory response of DCs co-cultured with HepG2 or Hep3B cell supernatant. Additionally, we have also observed an increase in immune-related cytokine secretion by DCs, including IL-6, IL-1β, and TNF-α, due to PSP treatment.
To our knowledge, PSP was reported to have anti-cancer effects, and its underlying mechanisms involve multiple molecules and signaling pathways. A previous study implied that PSP inhibited tumor growth and improved the spleen index, thymus index, cytokines secretion, and CD4+/CD8+ lymphocytes ratio through TLR4-MAPK/NF-κB signaling pathways.15) Polysaccharide-rich extract from Polygonatum sibiricum (PREPS) protect hematopoiesis inhibited by triple-negative breast cancer (TNBC) tumors in the bone marrow.28) PSP played a vital role in multiple myeloma (MM) by up-regulating the expression of p-phosphatidylinositol 3-kinase (PI3K), p-AKT, and p-mTOR.20) Our results demonstrated that PSP dose-dependently inhibited proliferation, migration, invasion, and EMT of liver cancer cells. Moreover, PSP dose-dependently reduced the activation of the TLR4/STAT3 signaling pathway in liver cancer cells.
Toll-like gene, a kind of protein molecule, played a very important role in non-specific immunity, and also serves as a link between specific and non-specific immunity.29) The TLR4 gene was an important part of the Toll-like gene family. TLR4 had a significant relationship with the occurrence and development of many tumors.30–32) A study showed that TLR4 activation mediated increased proliferation, and expression of the hepatomitogen epiregulin, which could be a therapeutic target for liver cancer prevention in advanced liver disease.33) Recently, studies showed that inhibition of TLR4-MyD88-NF-κB signaling suppressed hepatocyte tumorigenesis in vitro and in vivo.34) In addition, curcumin has been revealed to inhibit liver cancer by suppressing TLR4 signaling in HepG2 cells.35) Hepatitis B virus X (HBx) influenced the HCC progression by regulation of autophagy in response to TLR4 stimulation.36)
Furthermore, STAT3 was a latent cytoplasmic transcription factor that translocates to the nucleus following tyrosine phosphorylation at position 705 and dimerization.37) Activation of TLR4 signaling by LPS resulted in phosphorylation of STAT3.38) STAT3 activation was tightly controlled in healthy cells and was constitutively activated during tumorigenesis, where in it upregulated genes involved in tumor cell proliferation invasion, migration, and angiogenesis.39) STAT3 was overexpressed in approximately 60% of the liver cancer tissues and is associated with a poor prognosis.40) Consistent with this, blocking the STAT3 signaling pathway in human liver cancer cells using a decoy oligonucleotide induced apoptosis.41)
STAT3 functioned as a positive regulator to activate TGF-β1-induced EMT and metastasis of liver cancer.42) The STAT3 inhibitor, NSC 74859, induced liver cancer cell apoptosis and suppressed tumor growth.43) The ginsenoside compound K induced endoplasmic reticulum stress and apoptosis in Liver cancer cells by regulating STAT3.44) A previous study indicated that d targeting STAT3 signaling could interrupt the anti-apoptotic function of IL-6 in human liver cancer cells.45) Specifically, one report indicated that TLR4/STAT3 signaling pathway participated in the carcinogenesis, development, invasion, and metastasis of liver cancer.46) Hepatocellular carcinoma cells were inhibited by sorbafenib via suppression of TLR4/STAT3/SUMO1 pathway.47) TLR4/MyD88 activation up-regulated STAT3, which accelerated proliferation, metastasis, and multidrug resistance of liver cancer.48) The present study further demonstrated that stimulation of TLR4/STAT3 activity by LPS reversed the anti-oncogenic effects of PSP in liver cancer cells.
In conclusion, we demonstrated that PSP exerted its function as a tumor suppressor in liver cancer. Meanwhile, PSP promoted inflammation in the tumor microenvironment by inducing DCs maturation. Mechanistically, PSP significantly inhibited liver cancer progression by inhibiting TLR4/STAT3 signaling pathway. Thus, PSP may be useful as a potential therapeutic strategy for liver cancer in the future.
Author Contributions
Yunke Xu and Chuntao Li conceived and designed the experiments. Yunke Xu, Yong Guo, Changyou Lu, and Linlin Yu performed the experiments. Yunke Xu, Yong Guo, Changyou Lu, and Chao Fang analyzed the data. Chuntao Li contributed to the reagents and materials. Yunke Xu wrote the manuscript. Participate and publication consent was obtained from all authors.
Conflict of Interest
The authors declare no conflict of interest.
Data Availability
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- 1) Grandhi MS, Kim AK, Ronnekleiv-Kelly SM, Kamel IR, Ghasebeh MA, Pawlik TM. Hepatocellular carcinoma: from diagnosis to treatment. Surg. Oncol., 25, 74–85 (2016).
- 2) Chen Y, Tian Z. HBV-induced immune imbalance in the development of HCC. Front. Immunol., 10, 2048 (2019).
- 3) Nakade Y, Sato K, Nakao H, Yoneda M. Hepatocarcinogenesis in NASH. Gan To Kagaku Ryoho, 39, 693–697 (2012).
- 4) Sun B, Karin M. Inflammation and liver tumorigenesis. Front. Med., 7, 242–254 (2013).
- 5) Shang N, Bank T, Ding X, Breslin P, Li J, Shi B, Qiu W. Caspase-3 suppresses diethylnitrosamine-induced hepatocyte death, compensatory proliferation and hepatocarcinogenesis through inhibiting p38 activation. Cell Death Dis., 9, 558 (2018).
- 6) Aravalli RN, Cressman EN, Steer CJ. Cellular and molecular mechanisms of hepatocellular carcinoma: an update. Arch. Toxicol., 87, 227–247 (2013).
- 7) Couri T, Pillai A. Goals and targets for personalized therapy for HCC. Hepatol. Int., 13, 125–137 (2019).
- 8) Chen S, Cao Q, Wen W, Wang H. Targeted therapy for hepatocellular carcinoma: challenges and opportunities. Cancer Lett., 460, 1–9 (2019).
- 9) Fujiwara Y, Mangetsu M, Yang P, Kofujita H, Suzuki K, Ohfune Y, Shinada T. A quinone isolated from the nest of Vespa simillima and its growth-inhibitory effect on rat liver cancer cells. Biol. Pharm. Bull., 31, 722–725 (2008).
- 10) Yao J, Jiang Z, Duan W, Huang J, Zhang L, Hu L, He L, Li F, Xiao Y, Shu B, Liu C. Involvement of mitochondrial pathway in triptolide-induced cytotoxicity in human normal liver L-02 cells. Biol. Pharm. Bull., 31, 592–597 (2008).
- 11) Zhao P, Zhao C, Li X, Gao Q, Huang L, Xiao P, Gao W. The genus Polygonatum: a review of ethnopharmacology, phytochemistry and pharmacology. J. Ethnopharmacol., 214, 274–291 (2018).
- 12) Cui X, Wang S, Cao H, Guo H, Li Y, Xu F, Zheng M, Xi X, Han C. A review: the bioactivities and pharmacological applications of Polygonatum sibiricum polysaccharides. Molecules, 23, 1170 (2018).
- 13) Yelithao K, Surayot U, Lee JH, You S. RAW264.7 cell activating glucomannans extracted from rhizome of Polygonatum sibiricum. Prev. Nutr. Food Sci., 21, 245–254 (2016).
- 14) Wang S, Wang B, Hua W, Niu J, Dang K, Qiang Y, Wang Z. De novo assembly and analysis of Polygonatum sibiricum transcriptome and identification of genes involved in polysaccharide biosynthesis. Int. J. Mol. Sci., 18, 1950 (2017).
- 15) Long T, Liu Z, Shang J, Zhou X, Yu S, Tian H, Bao Y. Polygonatum sibiricum polysaccharides play anti-cancer effect through TLR4-MAPK/NF-κB signaling pathways. Int. J. Biol. Macromol., 111, 813–821 (2018).
- 16) Liu S, Jia QJ, Peng YQ, Feng TH, Hu ST, Dong JE, Liang ZS. Advances in mechanism research on Polygonatum in prevention and treatment of diabetes. Front. Pharmacol., 13, 758501 (2022).
- 17) Huang S, Yuan H, Li W, Liu X, Zhang X, Xiang D, Luo S. Polygonatum sibiricum polysaccharides protect against MPP-induced neurotoxicity via the Akt/mTOR and Nrf2 pathways. Oxid. Med. Cell. Longev., 2021, 8843899 (2021).
- 18) Han C, Sun T, Liu Y, Fan G, Zhang W, Liu C. Protective effect of Polygonatum sibiricum polysaccharides on gentamicin-induced acute kidney injury in rats via inhibiting p38 MAPK/ATF2 pathway. Int. J. Biol. Macromol., 151, 595–601 (2020).
- 19) Bu X, Liu Y, Wang L, Yan Z, Xin G, Su W. Oct4 promoted proliferation, migration, invasion, and epithelial-mesenchymal transition (EMT) in colon cancer cells by activating the SCF/c-Kit signaling pathway. Cell Cycle, 22, 291–302 (2023).
- 20) Zhao J, Ma L, Ni Z, Liu H. In vitro facilitating role of Polygonatum sibiricum polysaccharide in osteogenic differentiation of bone marrow mesenchymal stem cells from patients with multiple myeloma. Biotechnol. Lett., 43, 1311–1322 (2021).
- 21) Oura K, Morishita A, Tani J, Masaki T. Tumor immune microenvironment and immunosuppressive therapy in hepatocellular carcinoma: a review. Int. J. Mol. Sci., 22, 5801 (2021).
- 22) Xiang S, Li J, Shen J, Zhao Y, Wu X, Li M, Yang X, Kaboli PJ, Du F, Zheng Y, Wen Q, Cho CH, Yi T, Xiao Z. Identification of prognostic genes in the tumor microenvironment of hepatocellular carcinoma. Front. Immunol., 12, 653836 (2021).
- 23) Lin L, Chen S, Wang H, Gao B, Kallakury B, Bhuvaneshwar K, Cahn K, Gusev Y, Wang X, Wu Y, Marshall JL, Zhi X, He AR. SPTBN1 inhibits inflammatory responses and hepatocarcinogenesis via the stabilization of SOCS1 and downregulation of p65 in hepatocellular carcinoma. Theranostics, 11, 4232–4250 (2021).
- 24) Shi Y, Men X, Li X, Yang Z, Wen H. Research progress and clinical prospect of immunocytotherapy for the treatment of hepatocellular carcinoma. Int. Immunopharmacol., 82, 106351 (2020).
- 25) Zhang HM, Zhang LW, Liu WC, Cheng J, Si XM, Ren J. Comparative analysis of DC fused with tumor cells or transfected with tumor total RNA as potential cancer vaccines against hepatocellular carcinoma. Cytotherapy, 8, 580–588 (2006).
- 26) Shu G, Xu D, Zhao J, Yin L, Lin J, Fu H, Tang H, Fang J, Peng X, Zhao X. Protective effect of Polygonatum sibiricum polysaccharide on cyclophosphamide-induced immunosuppression in chickens. Res. Vet. Sci., 135, 96–105 (2021).
- 27) Liu Z, Ni H, Yu L, Xu S, Bo R, Qiu T, Gu P, Zhu T, He J, Wusiman A, Zhu S, Liu J, Hu Y, Wang D. Adjuvant activities of CTAB-modified Polygonatum sibiricum polysaccharide cubosomes on immune responses to ovalbumin in mice. Int. J. Biol. Macromol., 148, 793–801 (2020).
- 28) Xie Y, Jiang Z, Yang R, Ye Y, Pei L, Xiong S, Wang S, Wang L, Liu S. Polysaccharide-rich extract from Polygonatum sibiricum protects hematopoiesis in bone marrow suppressed by triple negative breast cancer. Biomed. Pharmacother., 137, 111338 (2021).
- 29) Li Y, Deng SL, Lian ZX, Yu K. Roles of toll-like receptors in nitroxidative stress in mammals. Cells, 8, 576 (2019).
- 30) Quirino MG, Macedo LC, Pagnano KBB, Pagliarini-e-Silva S, Sell AM, Visentainer JEL. Toll-like receptor gene polymorphisms in patients with myeloproliferative neoplasms. Mol. Biol. Rep., 48, 4995–5001 (2021).
- 31) Weng PH, Huang YL, Page JH, et al. Polymorphisms of an innate immune gene, toll-like receptor 4, and aggressive prostate cancer risk: a systematic review and meta-analysis. PLOS ONE, 9, e110569 (2014).
- 32) Semlali A, Jalouli M, Parine NR, Al Amri A, Arafah M, Al Naeem A, Abdullah Ajaj S, Rouabhia M, Alanazi MS. Toll-like receptor 4 as a predictor of clinical outcomes of estrogen receptor-negative breast cancer in Saudi women. Onco Targets Ther., 10, 1207–1216 (2017).
- 33) Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, Lefkowitch JH, Bower M, Friedman R, Sartor RB, Rabadan R, Schwabe RF. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell, 21, 504–516 (2012).
- 34) Hassan HM, Al-Wahaibi LH, Shehatou GS, El-Emam AA. Adamantane-linked isothiourea derivatives suppress the growth of experimental hepatocellular carcinoma via inhibition of TLR4-MyD88-NF-κB signaling. Am. J. Cancer Res., 11, 350–369 (2021).
- 35) Ren B, Luo S, Tian X, Jiang Z, Zou G, Xu F, Yin T, Huang Y, Liu J. Curcumin inhibits liver cancer by inhibiting DAMP molecule HSP70 and TLR4 signaling. Oncol. Rep., 40, 895–901 (2018).
- 36) Son J, Kim MJ, Lee JS, Kim JY, Chun E, Lee KY. Hepatitis B virus X protein promotes liver cancer progression through autophagy induction in response to TLR4 stimulation. Immune Netw., 21, e37 (2021).
- 37) Sehgal PB. Paradigm shifts in the cell biology of STAT signaling. Semin. Cell Dev. Biol., 19, 329–340 (2008).
- 38) Li X, Long J, He T, Belshaw R, Scott J. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci. Rep., 5, 12393 (2015).
- 39) Roos A, Dhruv HD, Peng S, Inge LJ, Tuncali S, Pineda M, Millard N, Mayo Z, Eschbacher JM, Loftus JC, Winkles JA, Tran NL. EGFRvIII-Stat5 signaling enhances glioblastoma cell migration and survival. Mol. Cancer Res., 16, 1185–1195 (2018).
- 40) He G, Yu GY, Temkin V, Ogata H, Kuntzen C, Sakurai T, Sieghart W, Peck-Radosavljevic M, Leffert HL, Karin M. Hepatocyte IKKbeta/NF-kappaB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell, 17, 286–297 (2010).
- 41) Chen H, Zhou B, Yang J, Ma X, Deng S, Huang Y, Wen Y, Yuan J, Yang X. Essential oil derived from Eupatorium adenophorum Spreng. mediates anticancer effect by inhibiting STAT3 and AKT activation to induce apoptosis in hepatocellular carcinoma. Front. Pharmacol., 9, 483 (2018).
- 42) Wang B, Liu T, Wu JC, Luo SZ, Chen R, Lu LG, Xu MY. STAT3 aggravates TGF-b1-induced hepatic epithelial-to-mesenchymal transition and migration. Biomed. Pharmacother., 98, 214–221 (2018).
- 43) Wu WY, Li J, Wu ZS, Zhang CL, Meng XL. STAT3 activation in monocytes accelerates liver cancer progression. BMC Cancer, 11, 506 (2011).
- 44) Zhang X, Zhang S, Sun Q, Jiao W, Yan Y, Zhang X. Compound K induces endoplasmic reticulum stress and apoptosis in human liver cancer cells by regulating STAT3. Molecules, 23, 1482 (2018).
- 45) Liu Y, Li PK, Li C, Lin J. Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. J. Biol. Chem., 285, 27429–27439 (2010).
- 46) Yao RR, Li JH, Zhang R, Chen RX, Wang YH. M2-polarized tumor-associated macrophages facilitated migration and epithelial-mesenchymal transition of HCC cells via the TLR4/STAT3 signaling pathway. World J. Surg. Oncol., 16, 9 (2018).
- 47) Li J, Zhou Y, Liu Y, Dai B, Zhang YH, Zhang PF, Shi XL. Sorafenib inhibits caspase-1 expression through suppressing TLR4/stat3/SUMO1 pathway in hepatocellular carcinoma. Cancer Biol. Ther., 19, 1057–1064 (2018).
- 48) Kang Y, Su G, Sun J, Zhang Y. Activation of the TLR4/MyD88 signaling pathway contributes to the development of human hepatocellular carcinoma via upregulation of IL-23 and IL-17A. Oncol. Lett., 15, 9647–9654 (2018).