Abstract
Most orally administered drugs exert their effects after being absorbed in the small intestine. Therefore, new drugs must undergo nonclinical pharmacokinetic evaluations in the small intestine. Enterocytes derived from human induced pluripotent stem cells (hiPSCs) are expected to be used in the evaluation system, as they reflect human intestinal characteristics more accurately; moreover, several differentiation protocols are available for these cells. However, enterocytes derived from hiPSCs have drawbacks such as time, cost, and lot-to-lot differences. Hence, to address these issues, we attempted to maintain hiPSC-derived intestinal stem cells (ISCs) that can differentiate into various intestinal cells by regulating various pathways. Although our previous attempt was partly successful, the drawbacks of elevated cost and complicated handling remained, because more than 10 factors (A 83-01, CHIR99021, epidermal growth factor, basic fibroblast growth factor, SB202190, nicotinamide, N-acetylcysteine, valproic acid, Wnt3a, R-spondin 1, and noggin) are needed to maintain ISCs. Therefore, in this study, we successfully maintained ISCs using only five factors, including growth factors. Moreover, we generated not only enterocytes but also intestinal organoids from the maintained ISCs. Thus, our novel findings provided a time-saving and cost-effective culture method for enterocytes derived from hiPSCs.
INTRODUCTION
Predicting accurate human pharmacokinetics at the nonclinical stage is essential to avoid drops of promising drug candidates in clinical trials and efficiently develop new drugs.1–3) In particular, pharmacokinetic-relevant studies in the small intestine are extremely important for predicting human pharmacokinetics and drug interactions because oral drugs are absorbed via the small intestine to exert their effects. In nonclinical studies, Caco-2 cells are used most frequently in pharmacokinetic studies of absorption. However, these cells have several disadvantages, such as a low expression of drug-metabolizing enzymes (e.g., CYP3A4), compared with human enterocytes.4–6) Although healthy human enterocytes are ideal for nonclinical pharmacokinetic studies, obtaining these cells regularly and stably is virtually impossible. Moreover, they would exhibit lot-to-lot differences even if they could be obtained.
Recently, the anticipation of human induced pluripotent stem cell (hiPSC)-derived enterocytes as materials to support drug discovery by resolving these issues has been increasing. hiPSC-derived enterocytes exhibit a phenotype that is more similar to that of human enterocytes than that of Caco-2 cells in terms of the expression patterns of drug-metabolizing enzymes and transporters7–9); furthermore, obtaining a stable supply is feasible. Although differentiation methods for hiPSC-derived enterocytes have been reported,8–13) these techniques exhibit several problems. First, the differentiation of hiPSCs to enterocytes is a long process. Human iPSCs are not readily available after the thawing of the cells, and researchers need to culture hiPSCs until the initiation of differentiation. In addition, the differentiation period is long (approximately 4 weeks) because hiPSC-derived enterocytes require maturation via endoderm cells and intestinal stem cells (ISCs).
The second problem is their cost, because differentiation into ISCs requires expensive factors, such as activin A. Furthermore, quality differences in hiPSC-derived enterocytes caused by differences among hiPSCs are also a problem. To address these issues, we expect to establish a maintenance culture method for hiPSC-derived ISCs, which are the progenitor cells of enterocytes.
In recent years, intestinal organoids have been attracting the attention of researchers. These organoids are cell aggregates composed of the various cells that make up the intestine, such as enterocytes, goblet cells, Paneth cells, and endocrine cells, as well as ISCs.14–16) Although the in vitro culture of ISCs is considered difficult, intestinal organoids allow it by reproducing their “niche” three-dimensionally, which is the microenvironment required for ISC survival.14,15) However, the number of ISCs in an intestinal organoid is limited to their use as an expansion culture of ISCs. In addition, ISCs need to be sorted when they are used for the differentiation to enterocytes, which is expected to be a strenuous task.
For these reasons, a long-term two-dimensional maintenance–culturing method is needed for ISCs. Despite the existence of several two-dimensional culture methods for ISCs, they are tissue-derived ISCs, not hiPSC-derived, and require a complicated procedure.17,18) We previously reported a method to maintain hiPSC-derived ISCs up to approximately three passages.19) However, this method requires improvement regarding labor and cost because of its requirement for more than 10 factors, including expensive proteins, such as Wnt3a and Noggin. Therefore, here, we developed a method that allows the maintenance culture of ISCs using fewer factors.
MATERIALS AND METHODS
Human iPSCsThe human iPSC line Windy was provided by Umezawa and colleagues at the National Center for Child Health and Development. Windy cells were cultured and maintained using our previously described method.10) The cells were cultured and maintained on mouse embryonic fibroblasts treated with mitomycin C (Kyowa Kirin Co., Ltd., Tokyo, Japan) in medium supplemented with 5 ng/mL of a basic fibroblast growth factor (FGF2) (GenScript, Piscataway, NJ, U.S.A.). The hiPSC line FF-1 was provided by FUJIFILM Corporation (Tokyo, Japan).
Differentiation and Maintenance of hiPSC-Derived ISCshiPSC-derived ISCs were differentiated for 7 d using our previously described method.19) For the first 3 d, hiPSCs were differentiated to endoderm cells by culturing in medium with 100 ng/mL activin A (PeproTech, Cranbury, NJ, U.S.A.), and endoderm cells were differentiated to ISCs by culturing in medium containing 250 ng/mL FGF2 for the remaining 4 d. The detailed medium composition is provided in Supplementary Table 1.
ISCs were maintained by changing the Advanced Dulbecco’s Modified Eagle Medium (DMEM)/F12 medium (Thermo Fisher Scientific, Waltham, MA, U.S.A.) containing 10% KnockOut Serum Replacement (Thermo Fisher Scientific), 1× penicillin–streptomycin solution (Biological Industries, Beit Haemek, Israel), 1% GlutaMax (Thermo Fisher Scientific), 100 ng/mL FGF2, 30 ng/mL epidermal growth factor (EGF) (GenScript), 2 µM Y-27632 (Focus Biomolecules, Plymouth Meeting, PA, U.S.A.), 0.5 µM A 83-01 (AdooQ Bioscience, Irvine, CA, U.S.A.), and 3 µM CHIR99021 (Focus Biomolecules) every 2 d on dishes coated with iMatrix-511 (0.25 µg/cm2) (Nippi, Tokyo, Japan). At 100% confluence, ISCs were dissociated using Accutase (Nacalai Tesque, Kyoto, Japan) and passaged on iMatrix-511-coated dishes.
Differentiation of Enterocytes from hiPSC-Derived ISCshiPSC-derived ISCs were differentiated to enterocytes using our previously described method.8) Windy-cell-derived ISCs were seeded on iMatrix-511 (0.25 µg/cm2)-coated plates or on Matrigel Growth Factor Reduced (GFR) (BD Biosciences, Bedford, MA, U.S.A.)-coated cell culture inserts (CORNING, Corning, NY, U.S.A.). FF-1-derived ISCs were seeded on Matrigel GFR-coated plates. These cells were cultured using medium containing 20 ng/mL EGF and 30 µM forskolin (FUJIFILM Wako, Osaka, Japan) for 7 d, followed by medium containing 20 ng/mL EGF, 30 µM forskolin, 20 µM PD98059 (AdooQ Bioscience), 5 µM 5-aza-2′-deoxycytidine (FUJIFILM Wako), and 0.5 µM A 83-01 for 12 d. The medium was changed every 3 d, and 10 µM Y-27632 was added to the medium only on the day of seeding.
Differentiation of hiPSC-Derived ISCs into Intestinal OrganoidsIntestinal organoids were differentiated using our previously described method.16) To generate intestinal organoids, hiPSC-derived ISCs were seeded on 60-mm EZSPHRE dishes (AGC Techno Glass Inc., Shizuoka, Japan) (1.0 × 106 cells/dish). hiPSC-derived ISCs were cultured in medium containing 100 ng/mL EGF, 100 ng/mL Noggin (GenScript), 200 ng/mL R-spondin 1 (GenScript), and 3% Matrigel GFR for 3 d. Thereafter, intestinal organoids were maintained on ultra-low-attachment dishes (BD Biosciences) for 24 d.
Freeze–Thawing of hiPSC-Derived ISCsThe pellets of hiPSC-derived ISCs were resuspended in CELLBANKER 1 (ZENOAQ, Fukushima, Japan) (1.5–2.0 × 106 cells/mL) and frozen at −80 °C, for storage. For thawing the cells, the frozen hiPSC-derived ISCs were semi-thawed at 37 °C in a water bath and completely dissolved in 10 mL of prewarmed medium. Subsequently, the cells were centrifuged at 100 × g for 5 min and resuspended in medium. Finally, the cells were seeded on the wells of plates for their differentiation to enterocytes.
Real-Time RT-Quantitative PCR (qPCR)Total RNA was extracted using the Agencourt RNAdvance Tissue Kit (Beckman Coulter, Brea, CA, U.S.A.), and cDNA was synthesized using the ReverTra Ace qPCR RT Master Mix (TOYOBO, Osaka, Japan) according to the manufacturer’s instructions. mRNA expression levels were detected using the primers listed in Table 1 and a KAPA SYBR Fast qPCR kit (F. Hoffmann-La Roche, Ltd., Basel, Switzerland) on a Light-Cycler® 96 System (F. Hoffmann-La Roche, Ltd.). The mRNA expression levels were normalized to that of the housekeeping gene hypoxanthine phosphoribosyltransferase 1 (HPRT1).
Table 1. Primers Used for Real-Time PCR Analysis
| Gene name | Sence (5′→3′) | Antisence (5′→3′) |
|---|
| ABCB1/MDR1 | CCCATCATTGCAATAGCAGG | TGTTCAAACTTCTGCTCCTGA |
| CDX2 | ACCTGTGCGAGTGGATGC | TCCTTTGCTCTGCGGTTCT |
| Chromogranin A | TCCGACACACTTTCCAAGCC | TTCTGCTGATGTGCCCTCTC |
| CYP3A4 | CTGTGTGTTTCCAAGAGAAGTTAC | TGCATCAATTTCCTCCTGCAG |
| EPHB2 | CACCTCGCTGCTTTATCTGGAGGAGCC | GAGGCTAGCTCACCTTCCACGGCG |
| HPRT | CTTTGCTTTCCTTGGTCAGG | TCAAGGGCATATCCTACAACA |
| ISX | CAGGAAGGAAGGAAGAGCAA | TGGGTAGTGGGTAAAGTGGAA |
| LGR5 | TGCTCTTCACCAACTGCATC | CTCAGGCTCACCAGATCCTC |
| Lysozyme | TCAATAGCCGCTACTGGTGT | AATGCCTTGTGGATCACGGA |
| MUC2 | AGAAGGCACCGTATATGACGAC | CAGCGTTACAGACACACTGCTC |
| SLC15A1/PEPT1 | CACCTCCTTGAAGAAGATGGCA | GGGAAGACTGGAAGAGTTTTATCG |
| Sucrase-isomaltase | GGTAAGGAGAAACCGGGAAG | GCACGTCGACCTATGGAAAT |
| Villin1 | AGCCAGATCACTGCTGAGGT | TGGACAGGTGTTCCTCCTTC |
| Vimentin | AGGAAATGGCTCGTCACCTTCGTGAATA | GGAGTGTCGGTTGTTAAGAACTAGAGCT |
hiPSC-derived ISCs and enterocytes on CellCarrier-96 microplates (PerkinElmer, Inc., Waltham, MA, U.S.A.) were washed with d-phosphate-buffered saline without Ca2+ and Mg2+ (d-PBS(−)), and then soaked with −30 °C methanol for 5 min at 25 °C, for fixation. After blocking in d-PBS(−) containing 5% fetal bovine serum (FBS) (Nichirei Biosciences Inc., Tokyo, Japan) for 20 min at room temperature (r.t.), the cells were incubated with primary antibodies at 4 °C overnight. Subsequently, the primary antibodies listed in Table 2 were reacted with Alexa Fluor 488- or 568-conjugated secondary antibodies (Thermo Fisher Scientific) for 1 h at r.t. under shading. The nuclei were stained with 4′,6-diamidino 2-phenylindole (DAPI; Dojindo, Kumamoto, Japan) (0.1 µg/mL) at r.t. for 5 min. The fluorescence images were captured and analyzed using the high-content imaging system Operetta (PerkinElmer, Inc.).
Table 2. Primary and Secondary Antibodies Used for Immunofluorescence Staining
| Antibodies | Source | Dilution |
|---|
| CDX | BioGenex | 1 : 50 |
| Chromogranin A | IMMUNOSTAR | 1 : 500 |
| Cytokeratin 20 | Abcam | 1 : 50 |
| E-cadherin | BD Transduction Laboratories | 1 : 100 |
| Ki67 | eBioscienc | 1 : 100 (ISC)/1 : 200 (organoid) |
| Lysozyme | BioGenex | — |
| MUC 2 | Santa Cruz Biotechnology | 1 : 50 (enterocyte)/1 : 200 (organoid) |
| Occludin | Santa Cruz Biotechnology | 1 : 100 |
| Olfactomedin 4 | Abcam | 1 : 100 |
| SOX9 | Merck | 1 : 100 |
| Villin | Santa Cruz Biotechnology | 1 : 50 |
| Alexa Fluor 488 Donkey Anti-Goat IgG (H&L) | Thermo Fisher Scientific | 1 : 200 |
| Alexa Fluor 488 Donkey Anti-Rabbit IgG (H&L) | Thermo Fisher Scientific | 1 : 200 |
| Alexa Fluor 488 Goat Anti-Mouse IgG (H&L) | Thermo Fisher Scientific | 1 : 200 |
| Alexa Fluor 568 Goat Anti-Mouse IgG (H&L) | Thermo Fisher Scientific | 1 : 200 |
ISC-derived intestinal organoids were embedded in Optimal Cutting Temperature Compound (Sakura Finetek Japan Co., Ltd., Tokyo, Japan) after fixing with 4% paraformaldehyde (FUJIFILM Wako) at 4 °C overnight. The embedded organoids were then cut into 10-µm sections using a Leica CM1950 instrument (Leica Microsystems, Wetzlar, Germany). The sections were activated with the ImmunoSaver antigen diluted to 1 : 200 (Nissin EM, Tokyo, Japan) at 98 °C for 45 min and permeabilized with 0.1% Triton X-100. After blocking in d-PBS(−) containing 5% FBS for 20 min, the sections were reacted with the primary antibodies, secondary antibodies, and DAPI using the method described above. The sections were mounted using the SlowFade® Diamond antifade reagent (Thermo Fisher Scientific), and confocal images were captured using a Zeiss LSM800 microscope and the AxioVision software (Carl Zeiss, Oberkochen, Germany).
Measurement of Transepithelial Electrical Resistance (TEER)Before measurements, cells were stood for 15 min and brought to r.t. The TEER values at the completion of the differentiation to enterocytes were measured using a Millicell ERS-2 V/ohm meter and a chopstick electrode (Merck Millipore, Burlington, MA, U.S.A.). The TEER values were calculated as follows:
The Liquid Chromatography (LC-MS/MS) Method for the Quantification of 1′-Hydroxy MidazolamPreparation of the Cell Culture SupernatantThe internal standard (IS) stock solution was prepared using 50% acetonitrile to 10 ng/mL. Standard solutions of 1′-hydroxy (1-OH) midazolam were prepared using transport buffer containing the IS stock solution at final concentrations of 0.3, 1, 3, 10, 30, 100, 300, and 1000 ng/mL, and kept at 4 °C before use.
Instruments and Analytical ConditionsThe 1-OH midazolam and IS were separated with an ACQUITY ultra performance liquid chromatography (UPLC) BEH C18 column (2.1 × 50 mm, 1.7 µm; Waters Corporation, Milford, MA, U.S.A.) at 40 °C, and measured using an AB SCIEX QTRAP 6500 LC-MS/MS system (AB SCIEX, Framingham, MA, U.S.A.). Ultrapure water containing 0.1% formic acid (solvent A) and methanol (solvent B) was used as the mobile phase for gradient elution, and the flow rate was set at 0.3 mL/min. The gradient condition of the mobile phase was as follows: 0–0.5 min 30% B, 0.5–2.0 min 30–50% B, 2.0–3.5 min 50–80% B, 3.5–3.6 min 80–95% B, 3.6–4.6 min 95% B, 4.6–4.8 min 95–30% B, and 4.8–6.0 min 30% B. The MS parameters were as follows: positive ion mode; 350 °C TIS temperature (TEM); 2000 V ionspray voltage; 20 psi curtain gas (CUR); 25 psi nebulizing gas (GS1); 60 psi TIS gas (GS2); 10 V entrance potential (EP); 95 V declustering potential (DP) and 30 eV collision energy (CE) for 1-OH midazolam and IS, respectively; and 18 V collision cell exit potential. The multiple reaction monitoring (MRM) transitions were m/z 341.9→324.0 and m/z 277.0→111.0 for 1-OH midazolam and IS, respectively. The data were analyzed using the MultiQuant software.
CYP3A4 ActivityThe cell pellets collected from the cell culture supernatant were lysed with 100 µL of Buffer RLT (QIAGEN, Venlo, the Netherlands) and centrifuged at 12000 rpm at 4 °C. Subsequently, the supernatants were collected and the protein concentration was measured using the BCA Protein Assay Kit (TaKaRa Bio Inc., Shiga, Japan). The value of 1-OH midazolam (pmol) was normalized to protein concentrations (mg protein) and time (h) as CYP3A4 activities.
RESULTS
Maintenance Culture of hiPSC-Derived ISCs Using Five FactorsGene expression in passaged hiPSC (Windy)-derived ISCs was measured using real-time RT-qPCR. Compared with passage 0 (P0), from P1, the gene expression levels of the leucine-rich repeat-containing G-protein-coupled receptor 5 (LGR5), which is an ISC marker, and of the caudal type homeobox 2 (CDX2), which is a midgut/hindgut marker, were decreased and increased, respectively. However, the mRNA expression levels of both LGR5 and CDX2 were almost constant beyond P1 (Fig. 1A). In addition, the gene expression pattern in FF-1-derived ISCs was similar (Supplementary Fig. 1). The CDX2, Sry-related HMG box transcription factor9 (SOX9; which is an intestinal progenitor cell marker), and Ki67 (which is a cell proliferation marker) proteins were similarly expressed between P1 and P3 (Fig. 1B). Furthermore, the number of cells from P0 to P1 was set as 1, and the relative cell proliferation ratio was calculated by counting the number of cells in each passage after P1. Constant cell proliferation was observed, and the proliferation ratio remained almost unchanged, even if the number of culture days increased and the number of passages accumulated. The approximate curve correlation coefficient (R2) was 0.9914. The number of ISCs on day 25 (P7) was approximately 150 times greater than that recorded on day 7 (P1), and the doubling time was calculated to be 2.3 d (Fig. 1C).
Differentiation of ISCs Maintained with Five Factors into Enterocytes at Different PassagesEnterocytes were differentiated from hiPSC-derived ISCs maintained with five factors at different passages, and the gene expression of enterocyte markers was measured. The gene expression of enterocyte markers (villin1, sucrase-isomaltase, and intestine-specific homeobox (ISX)) and drug transporters (solute carrier family 15 member 1/Peptide transporter 1 (SLC15A1/PEPT1) and ATP-binding cassette subfamily B member 1/multiple drug resistance 1 (ABCB1/MDR1)) in the small intestine from P1 was similar compared with that observed at P0. However, the expression of the gene encoding CYP3A4, which is a drug-metabolizing enzyme, at P4 was less than 10% of that detected at P0 (Fig. 2A). It was also confirmed that enterocytes at all passages expressed essentially the same mRNA levels as those observed at P0, even if FF-1, which are a different hiPSC line from Windy, were used (Supplementary Fig. 2A). Expression of the villin and cytokeratin 20 proteins, which are enterocyte markers, and of mucin 2 (MUC2), which is a goblet cell marker, was observed at different passages (Fig. 2B). Regarding the enterocytes derived from FF-1 ISCs at P1, P3, and P5, cells at each passage expressed the villin protein regardless of the passage number, similar to that observed for differentiation from Windy (Supplementary Fig. 2B).
TEER and CYP3A4 metabolic activity were measured as a functional evaluation of hiPSC-derived enterocytes. The values of TEER, which reflects tight junction formation, at P1, P2, and P3 on day 26 were equal to or larger than those detected at P0 (Fig. 3A). Moreover, 1-OH midazolam, which is a metabolite of midazolam via CYP3A4, was measured to assess the metabolic activity of CYP3A4. As a consequence, 1-OH midazolam was detected in all enterocytes up to P3, regardless of the number of passages (Fig. 3B).
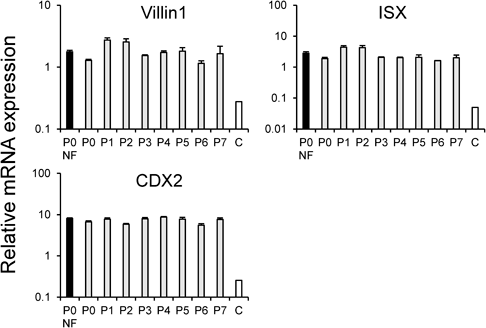
Freeze–Thawing of hiPSC-Derived ISCsNext, we investigated whether ISCs derived from hiPSCs could be stored frozen. After freezing and storing each passage of ISCs derived from hiPSCs, the cells were thawed, seeded, and differentiated; finally, an mRNA expression analysis was performed. It was confirmed that the mRNA expression levels of villin1, ISX, and CDX2 remained virtually unchanged compared with those of the enterocytes that were differentiated from P0 ISCs without freezing (Fig. 4).
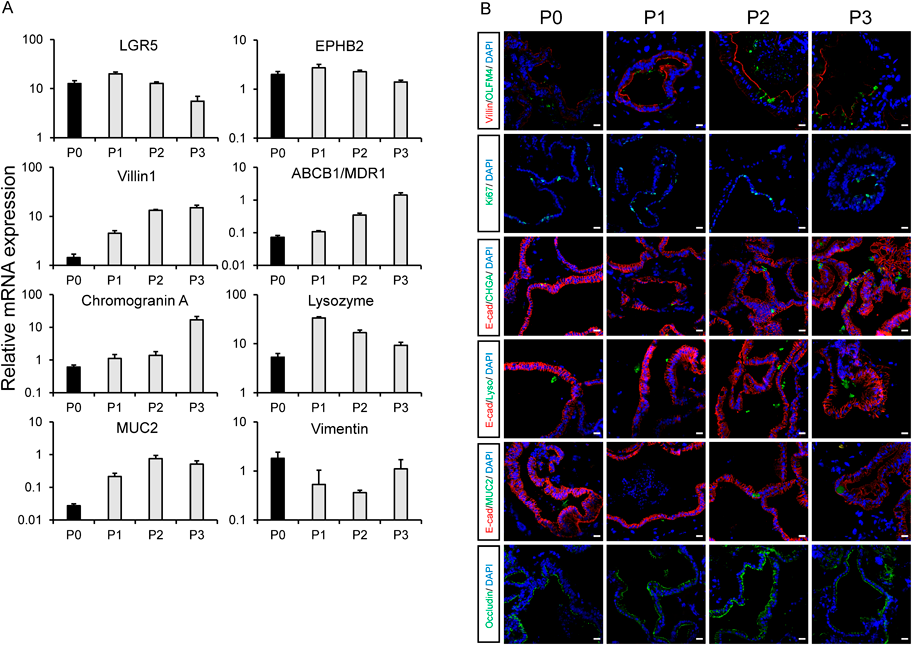
Generation of Intestinal Organoids from hiPSC-Derived ISCsBubble-like intestinal organoids could be constructed from hiPSC-derived ISCs at all passages (Figs. 5A–F). The expression levels of the following marker genes were analyzed: LGR5, ephrin receptor B2 (EPHB2, as an ISC marker), villin1, ABCB1/MDR1, chromogranin A (as an intestinal endocrine marker), lysozyme (as Paneth cell marker), MUC2 (as a goblet cell marker), and vimentin (as a mesenchymal cell marker). Furthermore, the expression of the LGR5, EPHB2, and vimentin genes at P1 to P3 was similar to that recorded at P0, whereas the levels of the villin, ABCB1/MDR1, chromogranin A, lysozyme, and MUC2 genes after P1 tended to be higher than those observed at P0 (Fig. 6A).
Next, immunofluorescence staining of the intestinal organoids generated from each passage of ISCs was performed. At all passages, the expression of the following proteins was confirmed: the ISC marker olfactomedin 4 (OLFM4); villin; Ki67; cell-adhesion molecule E-cadherin; chromogranin A; lysozyme; and the tight junction marker occludin. Villin and occludin expression was observed on the apical side of the intestinal organoids (Fig. 6B). Moreover, the expression of the small intestine marker gene apolipoprotein B (APOB) in the intestinal organoids was as high as that detected in the human small intestine and higher than that detected in the human colon. Conversely, the colon marker gene homeobox A3 (HOXA3) exhibited a similar expression level to that of the human small intestine and was lower than that of the human colon (Supplementary Fig. 3).
DISCUSSION
In this study, we attempted to reduce the number of factors that are required to maintain ISCs. The analysis of the gene expression patterns of ISCs maintained with Y-27632, A83-01, CHIR99021, and growth factors (EGF and FGF2) revealed that the expression of the LGR5 and CDX2 genes remained nearly constant in ISCs at all passages (Fig. 1A). Because LGR5 is a small intestine stem cell marker20) and CDX2 is a midgut/hindgut marker,21) this result suggests that hiPSC-derived ISCs can be cultured. In contrast, the expression level of CDX2 at P0 was lower than that detected in the adult human small intestine, and that recorded after P1 was increased compared with that observed after P0. Therefore, it is possible that the differentiation of hiPSCs to ISCs was not uniform on day 7 (P0), and ISCs were selected from P0 to P1 passages. One reason for this selection was the contribution of iMatrix-511, which is used as a coating material. It is known that laminin 511 is expressed at high levels in the small intestine,22) and that iMatrix-511, which is an E8 fragment of laminin 511, has a binding affinity to α6β1 integrin that is similar to that of full-length laminin 511.23) Therefore, only cells that successfully differentiated into ISCs may have selectively attached to the culture dishes. The expression level of LGR5 after P1 was lower than that observed after P0. However, ISCs may have been maintained in culture because they retained the characteristics of ISCs, as the expression of the LGR5 and CDX2 genes after P1 was higher than that of the adult small intestine.
In the small intestine, cell turnover occurs constantly, and ISCs are the source of all cells in the small intestine. Immunofluorescence staining of ISCs at P1 and P3 revealed the expression of the cell proliferation maker Ki67, in addition to SOX9 and CDX2 (Fig. 1B). We also confirmed that the cell proliferation rate remained nearly constant up to day 25 (Fig. 1C). This suggests that ISCs could be maintained in culture with cell proliferation ability. Although SOX9, which is an ISC and intestinal progenitor cell marker, is expressed in crypt parts of the small intestine, its specificity as an ISC marker is relatively lower than that of LGR5.24) Therefore, as reported by Sato et al. by incorporating a green fluorescent protein under the control of the LGR5 promoter into hiPSCs,15) it is considered that a more-accurate and real-time evaluation of ISC culture availability can be carried out by visualizing LGR5 protein expression and confirming the LGR5-positive cell ratio in a timely manner.
hiPSC-derived enterocytes are expected to be used as an evaluation tool instead of Caco-2 cells. Therefore, as the subsequent step of this investigation, we differentiated the maintained ISCs to enterocytes. The expression of several enterocyte markers was confirmed based not only based on gene expression, but also on protein expression (Fig. 2). Accordingly, we consider that enterocytes up to P7 could be differentiated from the maintained ISCs using the protocol proposed here. Furthermore, we performed functional evaluations of enterocytes differentiated from ISCs (Fig. 3). Because P1–P3 enterocytes on day 26 exhibited TEER values that were equal to or greater than those detected at P0, we considered that a tight junction was formed even if enterocytes were differentiated from ISCs after repeated passages. In addition to the TEER value, an equivalent CYP3A4 metabolic activity was confirmed in P1–P3 enterocytes compared with that recorded at P0. Consequently, we propose that enterocytes derived from ISC maintained using the method described in this study may be a beneficial evaluation tool instead of Caco-2 cells, as hiPSC-derived enterocytes reported so far.9)
Considering their usefulness and convenience in drug discovery research, it is remarkably important that ISCs derived from hiPSCs have the ability to be frozen and thawed. Therefore, we froze maintenance-cultured ISCs at each passage, and the cells were thawed and differentiated into enterocytes after storage for a few days (Fig. 4). A gene expression analysis confirmed that the levels of several enterocyte markers were the same as those detected after direct differentiation without freezing. This result suggests that maintained hiPSCs-derived ISCs can be frozen and stored in a status amenable to differentiation to enterocytes.
Finally, we attempted to differentiate hiPSC-derived ISCs into intestinal organoids. Differentiated intestinal organoids exhibited shapes similar to those reported previously; they were bubble-like and contained various types of intestinal cells, such as intestinal endocrine cells, Paneth cells, goblet cells, and ISCs16) (Figs. 5, 6). Moreover, given the nearly same gene expression level of intestinal cell markers as adult small intestine markers in organoids derived from each passage ISC, maintenance-cultured ISCs appear to have the ability to differentiate into the various cell types that compose the small intestine, regardless of the number of passages. While ISC markers do not show a change through P0 to P3, the gene expression of intestinal markers, such as Villin1, tends to increase after P1; thus, more intestine-like organoids maintaining ISCs may be formed after P1 than after P0. This could be due to the inhibition of differentiation into cells, except intestine linage by ISC selection by passage, as stated in the discussion of Fig. 1.
For the maintenance of ISCs, is necessary to reproduce their “niche” in vitro. Wnt signaling, which is considered to be extremely important in the niche of ISCs,25) is activated by the GSK-3β inhibitory effect of CHIR99021. In addition, passages are an essential process for two-dimensional maintenance culture; however, the passage procedure causes damage to cells. Because it is known that epithelial–mesenchymal transition (EMT) is triggered by inflammation via transforming growth factor β (TGF-β),26) it is possible that A 83-01 suppresses the differentiation of ISCs to mesenchymal cells by inhibiting TGF-β. Furthermore, as it has been reported that the number of colon stem cells tends to increase after inhibiting TGF-β,27) we also propose that the inhibition of TGF-β is important for the maintenance of gastrointestinal stem cells. Moreover, when pluripotent stem cells are passaged, Y-27632 is generally used to suppress cell death via Rho-associated protein kinase (ROCK) inhibition.28) Hence, we suggest that the addition of Y-27632 contributed to the suppression of cell death because of the damage caused by cell passaging during maintenance culture and cell proliferation. Furthermore, EGF was usually employed in previously reported intestinal organoid culture protocols,15,16) and the FGF2 and insulin-like growth factor 1 (IGF-1) are needed for intestinal organoid self-proliferation.29) In addition, ISCs could not be maintained and increased in maintenance culture without the addition of EGF and FGF2 (data not shown). Therefore, in the present protocol, it is considered that the reproduction of the ISC niche in vitro using EGF, FGF2, and CHIR99021; the mitigation of cell damage by A 83-01 and Y-27632; and the promotion of cell proliferation by EGF, FGF2, and Y-27632 resulted in the achievement of a two-dimensional maintenance culture of hiPSC-derived ISCs using only five factors.
Rat mature hepatocytes were reprogrammed into rat hepatocyte progenitor cells and their culture could be maintained two-dimensionally using three small molecules, i.e., Y-27632, A 83-01, and CHIR99021.30) We obtained a similar result here, probably because both the liver and intestine are endoderm-derived organs.
In this study, we successfully differentiated ISCs into enterocytes up to passage 7 on cell culture plates. However, it was sometimes difficult to evaluate differentiation on cell culture inserts because ISCs were dissociated after P4. In intestinal organoid culture or endoderm cell culture in vitro, some reports have shown that bone morphogenetic protein (BMP) signal inhibition or growth factors other than EGF and FGF2 are essential, in addition to Wnt signaling.27,29,31) Therefore, by adding factors that reproduce the ISC “niche” more accurately, it may be possible to perform maintenance culture for a longer period and induce differentiation into enterocytes more stably.
We concluded that ISCs were maintained using only five factors (Y-27632, A83-01, CHIR99021, EGF, and FGF2), which is less than half of the previously reported factors. The application of hiPSC-derived ISCs maintained using the protocol developed in the present study should reduce the cost and effort during maintenance culture compared with previously reported methods; moreover, it will reduce the cost and time required to generate hiPSC-derived enterocytes. Finally, this tool is expected to produce many homogeneous intestinal organoids that can be used for drug-toxicity evaluation.
Acknowledgments
The authors express our sincere thanks to Dr. Hidenori Akutsu, Dr. Yoshitaka Miyagawa, Dr. Hajime Okita, Dr. Nobutaka Kiyokawa, Dr. Masashi Toyoda, and Dr. Akihiro Umezawa for providing hiPS cell line, Windy. We would like to thank FUJIFILM Corporation for providing hiPS cell line, FF-1. This work was supported by a Grant-in-Aid from the Japan Agency for Medical Research and Development (AMED) Grant Numbers: JP21be0304203, JP22be1004101, JP23be1004101, JSPS KAKENHI Grant Numbers: JP15K18929 and JP22H02738, the Research Foundation for Pharmaceutical Sciences and FUJIFILM Corporation.
Conflict of Interest
Shota Mizuno is an employee of Daiichi Sankyo Co., Ltd. The other authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
REFERENCES
- 1) Dickson M, Gagnon JP. The cost of new drug discovery and development. Discov. Med., 4, 172–179 (2009).
- 2) Kaitin KI. Deconstructing the drug development process: the new face of innovation. Clin. Pharmacol. Ther., 87, 356–361 (2010).
- 3) Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov., 9, 203–214 (2010).
- 4) Harwood MD, Achour B, Neuhoff S, Russell MR, Carlson G, Warhurst G, Rostami-Hodjegan A. In vitro-in vivo extrapolation scaling factors for intestinal P-glycoprotein and breast cancer resistance protein: Part I: a cross-laboratory comparison of transporter-protein abundances and relative expression factors in human intestine and Caco-2 cells. Drug Metab. Dispos., 44, 297–307 (2016).
- 5) Sun D, Lennernas H, Welage LS, Barnett JL, Landowski CP, Foster D, Fleisher D, Lee KD, Amidon GL. Comparison of human duodenum and Caco-2 gene expression profiles for 12,000 gene sequences tags and correlation with permeability of 26 drugs. Pharm. Res., 19, 1400–1416 (2002).
- 6) Nakamura T, Sakaeda T, Ohmoto N, Tamura T, Aoyama N, Shirakawa T, Kamigaki T, Nakamura T, Kim KI, Kim SR, Kuroda Y, Matsuo M, Kasuga M, Okumura K. Real-time quantitative polymerase chain reaction for MDR1, MRP1, MRP2, and CYP3A-mRNA levels in Caco-2 cell lines, human duodenal enterocytes, normal colorectal tissues, and colorectal adenocarcinomas. Drug Metab. Dispos., 30, 4–6 (2002).
- 7) Kabeya T, Matsumura W, Iwao T, Hosokawa M, Matsunaga T. Functional analysis of carboxylesterase in human induced pluripotent stem cell-derived enterocytes. Biochem. Biophys. Res. Commun., 486, 143–148 (2017).
- 8) Kabeya T, Mima S, Imakura Y, Miyashita T, Ogura I, Yamada T, Yasujima T, Yuasa H, Iwao T, Matsunaga T. Pharmacokinetic functions of human induced pluripotent stem cell-derived small intestinal epithelial cells. Drug Metab. Pharmacokinet., 35, 374–382 (2020).
- 9) Kabeya T, Qiu S, Hibino M, Nagasaki M, Kodama N, Iwao T, Matsunaga T. Cyclic AMP signaling promotes the differentiation of human induced pluripotent stem cells into intestinal epithelial cells. Drug Metab. Dispos., 46, 1411–1419 (2018).
- 10) Iwao T, Toyota M, Miyagawa Y, Okita H, Kiyokawa N, Akutsu H, Umezawa A, Nagata K, Matsunaga T. Differentiation of human induced pluripotent stem cells into functional enterocyte-like cells using a simple method. Drug Metab. Pharmacokinet., 29, 44–51 (2014).
- 11) Iwao T, Kodama N, Kondo Y, Kabeya T, Nakamura K, Horikawa T, Niwa T, Kurose K, Matsunaga T. Generation of enterocyte-like cells with pharmacokinetic functions from human induced pluripotent stem cells using small-molecule compounds. Drug Metab. Dispos., 43, 603–610 (2015).
- 12) Ogaki S, Shiraki N, Kume K, Kume S. Wnt and Notch signals guide embryonic stem cell differentiation into the intestinal lineages. Stem Cells, 31, 1086–1096 (2013).
- 13) Negoro R, Takayama K, Kawai K, Harada K, Sakurai F, Hirata K, Mizuguchi H. Efficient generation of small intestinal epithelial-like cells from human iPSCs for drug absorption and metabolism studies. Stem Cell Reports, 11, 1539–1550 (2018).
- 14) Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, Shroyer NF, Wells JM. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature, 470, 105–109 (2011).
- 15) Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature, 459, 262–265 (2009).
- 16) Onozato D, Yamashita M, Nakanishi A, Akagawa T, Kida Y, Ogawa I, Hashita T, Iwao T, Matsunaga T. Generation of intestinal organoids suitable for pharmacokinetic studies from human induced pluripotent stem cells. Drug Metab. Dispos., 46, 1572–1580 (2018).
- 17) Scott A, Rouch JD, Jabaji Z, Khalil HA, Solorzano S, Lewis M, Martín MG, Stelzner MG, Dunn JC. Long-term renewable human intestinal epithelial stem cells as monolayers: a potential for clinical use. J. Pediatr. Surg., 51, 995–1000 (2016).
- 18) Tong Z, Martyn K, Yang A, Yin X, Mead BE, Joshi N, Sherman NE, Langer RS, Karp JM. Towards a defined ECM and small molecule based monolayer culture system for the expansion of mouse and human intestinal stem cells. Biomaterials, 154, 60–73 (2018).
- 19) Kondo S, Mizuno S, Hashita T, Iwao T, Matsunaga T. Establishment of a novel culture method for maintaining intestinal stem cells derived from human induced pluripotent stem cells. Biol. Open, 9, bio049064 (2020).
- 20) Barker N, van Es JH, Kuipers J, Kujala P, van den Born M, Cozijnsen M, Haegebarth A, Korving J, Begthel H, Peters PJ, Clevers H. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature, 449, 1003–1007 (2007).
- 21) Dye BR, Hill DR, Ferguson MA, Tsai YH, Nagy MS, Dyal R, Wells JM, Mayhew CN, Nattiv R, Klein OD, White ES, Deutsch GH, Spence JR. In vitro generation of human pluripotent stem cell derived lung organoids. eLife, 4, e05098 (2015).
- 22) Ritié L, Spenlé C, Lacroute J, Bolcato-Bellemin AL, Lefebvre O, Bole-Feysot C, Jost B, Klein A, Arnold C, Kedinger M, Bagnard D, Orend G, Simon-Assmann P. Abnormal Wnt and PI3Kinase signaling in the malformed intestine of lama5 deficient mice. PLOS ONE, 7, e37710 (2012).
- 23) Taniguchi Y, Ido H, Sanzen N, Hayashi M, Sato-Nishiuchi R, Futaki S, Sekiguchi K. The C-terminal region of laminin beta chains modulates the integrin binding affinities of laminins. J. Biol. Chem., 284, 7820–7831 (2009).
- 24) Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, Kuhara T, Hosokawa S, Elbahrawy A, Soeda T, Koizumi M, Masui T, Kawaguchi M, Takaori K, Doi R, Nishi E, Kakinoki R, Deng JM, Behringer RR, Nakamura T, Uemoto S. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat. Genet., 43, 34–41 (2011).
- 25) Fevr T, Robine S, Louvard D, Huelsken J. Wnt/beta-catenin is essential for intestinal homeostasis and maintenance of intestinal stem cells. Mol. Cell. Biol., 27, 7551–7559 (2007).
- 26) Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res., 19, 156–172 (2009).
- 27) Reynolds A, Wharton N, Parris A, et al. Canonical Wnt signals combined with suppressed TGFβ/BMP pathways promote renewal of the native human colonic epithelium. Gut, 63, 610–621 (2014).
- 28) Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K, Sasai Y. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nat. Biotechnol., 25, 681–686 (2007).
- 29) Fujii M, Matano M, Toshimitsu K, Takano A, Mikami Y, Nishikori S, Sugimoto S, Sato T. Human intestinal organoids maintain self-renewal capacity and cellular diversity in niche-inspired culture condition. Cell Stem Cell, 23, 787–793.e6 (2018).
- 30) Katsuda T, Kawamata M, Hagiwara K, Takahashi R, Yamamoto Y, Camargo FD, Ochiya T. Conversion of terminally committed hepatocytes to culturable bipotent progenitor cells with regenerative capacity. Cell Stem Cell, 20, 41–55 (2017).
- 31) Zhang RR, Koido M, Tadokoro T, Ouchi R, Matsuno T, Ueno Y, Sekine K, Takebe T, Taniguchi H. Human iPSC-derived posterior gut progenitors are expandable and capable of forming gut and liver organoids. Stem Cell Reports, 10, 780–793 (2018).