Abstract
Traumatic brain injury (TBI) is severe damage to the head caused by traffic accidents, falls, and sports. Because TBI-induced disruption of the blood–brain barrier (BBB) causes brain edema and neuroinflammation, which are major causes of death or serious disabilities, protection and recovery of BBB function may be beneficial therapeutic strategies for TBI. Astrocytes are key components of BBB integrity, and astrocyte-derived bioactive factors promote and suppress BBB disruption in TBI. Therefore, the regulation of astrocyte function is essential for BBB protection. In the injured cerebrum of TBI model mice, we found that the endothelin ETB receptor, histamine H2 receptor, and transient receptor potential vanilloid 4 (TRPV4) were predominantly expressed in reactive astrocytes. We also showed that repeated administration of an ETB receptor antagonist, H2 receptor agonist, and TRPV4 antagonist alleviated BBB disruption and brain edema in a TBI mouse model. Furthermore, these drugs decreased the expression levels of astrocyte-derived factors promoting BBB disruption and increased the expression levels of astrocyte-derived protective factors in the injured cerebrum after TBI. These results suggest that the ETB receptor, H2 receptor, and TRPV4 are molecules that regulate astrocyte function, and might be attractive candidates for the development of therapeutic drugs for TBI.
1. INTRODUCTION
Traumatic brain injury (TBI) is severe damage to the head resulting from an external force caused by traffic accidents, falls, sports, etc. TBI is a major cause of unexpected death in young and old people worldwide and results in serious disabilities of motor, sensory, mental, and cognitive functions that cause an impressive decline in the QOL of surviving patients. Although emergent treatments are essential, current treatments such as decompressive craniotomy, hyperosmolar treatment, and sedation are extremely limited, and beneficial therapeutic drugs have not yet been developed. In the acute phase of TBI, the function of the blood–brain barrier (BBB) is substantially impaired by decreasing tight junction proteins and the loss of cells comprising the BBB,1,2) and disruption of the BBB causes brain edema and neuroinflammation, which are major causes of unexpected death or serious disabilities.3–5) Brain edema, especially vasogenic edema, results from the extravasation of intravascular water and serum proteins into the cerebral parenchyma following increased intracranial pressure and hernia, which causes critical impairment of the central nervous system and often results in the death of patients with TBI6,7) (Fig. 1). Neuroinflammation results from the infiltration of intravascular neutrophils and immune cells into the cerebral parenchyma, which exacerbates neuronal damage and causes various neurological dysfunctions in TBI patients.8,9) Therefore, the protection and recovery of BBB function are beneficial and essential therapeutic strategies for TBI.
Astrocytes are the most abundant type of glial cell in the brain and are key components of the BBB, together with endothelial cells and pericytes. Astrocytic endfeet surround the cerebral microvessels, which strictly control the barrier function of the BBB. Additionally, astrocytes release various bioactive factors that regulate BBB permeability under both physiological and pathophysiological conditions.10) In response to brain damage, astrocytes transform from the resting to the reactive state, which is characterized by increased glial fibrillary acidic protein (GFAP) expression, hypertrophy, and ability to proliferate, resulting in astrogliosis.11,12) Reactive astrocytes exert protective and detrimental effects against brain damage through bioactive factors.10) Some studies have suggested a relationship between TBI and reactive astrocytes in both patients and experimental animal models. In patients with TBI, phenotypic conversion to reactive astrocytes was predominantly observed in the damaged areas.13) In experimental TBI animal models, the number of reactive astrocytes increased in the injured cerebrum.1,14,15) These findings suggest that astrocytes are involved in the pathogenesis of TBI. Therefore, we focused on the functional astrocytic molecules to identify novel therapeutic drugs for TBI.
2. METHODS FOR EVALUATING EXPERIMENTAL TBI
2.1. Fluid Percussion Injury (FPI)FPI is an established experimental model of TBI performed by hydraulic impact on the dura mater of experimental animals using a fluid percussion device (AmScien Instruments, Richmond, VA, U.S.A.; model FP302) (Fig. 2A). An experimental animal is placed in a stereotactic device under anesthesia, and the skull is exposed. A 3-mm-diameter hole is drilled in the skull of the left hemisphere, and a lure fitting (ISIS Co., Ltd., Osaka, Japan; Cat. No. VRS306) (Fig. 2A) is fixed to the drilled area. The interior of the lure fitting is filled with sterilized saline, and the lure fitting is connected to a tube of the fluid percussion device, after which a hydraulic insult is applied in the range 1.2 to 1.4 atmospheres.1)
FPI is also performed on cultured cells using a cell trauma chamber and fluid percussion device (Fig. 2B). Cultured astrocytes are prepared from mouse cerebrum and incubated in 35-mm culture dishes (Corning, Corning, NY, U.S.A.; Cat. No. 430165). Most cultured cells are GFAP-positive (Fig. 3A). The culture dish is placed in the cell trauma chamber of the FPI device (AmScien Instruments, model FP302) (Fig. 2B). After the upper half of the chamber is filled with sterilized saline, the central tube of the chamber is connected to the fluid percussion device, and FPI is performed on cultured astrocytes.16)
2.2. Evaluation of BBB Disruption by Extravasation of Evans Blue or Fluorescein Isothiocyanate (FITC)-DextranBBB disruption is determined by extravasation of Evans blue (Sigma-Aldrich, St. Louis, MO, U.S.A.; Cat. No. E2129-10G) or FITC-dextran (Sigma-Aldrich, Cat. No. FD4-100MG) into the cerebral parenchyma.16) Saline containing 4% Evans blue or 4% FITC-dextran is administered via the tail vein at 3 mL/kg. Sixty minutes after administration, an aliquot of circulatory blood is collected to determine serum Evans blue or FITC-dextran content. The experimental animals are intracardially perfused with 50 mL of phosphate-buffered saline under anesthesia. The perfused cerebrum is collected, and a 5-mm thick coronal brain section (between 0 and 5 mm posterior to the bregma) is prepared using a microtome. The prepared brain tissues are weighed and immersed in 400 µL formamide (Wako Pure Chemical Corporation, Osaka, Japan; Cat. No. 065-00436) at 55 °C overnight. Evans blue content in the extract is determined by measuring the absorbance at 655 nm, and FITC-dextran content is determined by measuring the fluorescence intensity at 528 nm with 485 nm excitation. The percentage of Evans blue or FITC-dextran extravasation in brain tissue is calculated using the following formula:
2.3. Determination of Brain Edema by Measuring the Brain Water ContentBrain edema is determined based on increased brain water content.1) After FPI, the mouse brain tissue is collected and a 5-mm thick coronal section (between 0 and 5 mm posterior to the bregma) is prepared using a microtome. The prepared brain tissues are weighed (wet weight), dried at 70 °C overnight, and then dried tissues are weighed (dry weight). The percentage brain water content is calculated using the following formula:
The brain water content of normal brain tissue is approximately 76 to 77%, whereas that of damaged brain tissue is over 80%.
3. ROLES OF ASTROCYTES IN THE DISRUPTION AND RECOVERY OF THE BBB IN TBI
In the damaged brain, reactive astrocytes produce various bioactive factors including vascular permeability-accelerating factors and protective factors that regulate permeability of the BBB.10) Factors that accelerate vascular permeability include endothelin-1 (ET-1), matrix metalloproteinase-9 (MMP-9), and vascular endothelial growth factor-A (VEGF-A), which promote BBB permeability and exacerbate BBB disruption in damaged brain.1,17–20) The vascular protective factors include angiopoietin-1 (Ang-1) and sonic hedgehog (Shh), which decrease BBB permeability and alleviate BBB disruption.16,21–26) In contrast, vascular permeability-accelerating factors decrease the expression levels of endothelial tight junction proteins, including claudin-5 and occludin, which are essential components for the barrier function of the BBB,27–31) whereas vascular protective factors increase these expression levels.16,22,23,32,33)
After FPI, the number of GFAP-positive reactive astrocytes increases in injured mouse cerebrum (Figs. 3B, C). Moreover, the expression of vascular permeability-accelerating factors, including MMP-9 and VEGF-A, are predominantly observed in GFAP-positive reactive astrocytes (Fig. 4A). In addition, the expression levels of MMP-9 and VEGF-A are increased in cultured astrocytes following in vitro FPI.34) Repeated intracerebroventricular (i.c.v.) administration of an MMP-9 inhibitor or VEGF-neutralizing antibody alleviates BBB disruption in experimental TBI model mice,1,31) and astrocyte-derived MMP-9 and VEGF-A promote TBI-induced BBB disruption.
In the injured cerebrum after FPI, the expression of the vascular protective factor Ang-1 is predominantly observed in GFAP-positive reactive astrocytes in the injured cerebrum after FPI (Fig. 4A). Similarly, Shh expression is also observed in astrocytes in the injured cerebrum after FPI,26) and FPI increases the expression of Shh in cultured astrocytes (Fig. 4B). Repeated i.c.v. administration of recombinant Ang-1 or Shh reduces TBI-induced BBB disruption in experimental mice.16,26) These results suggest that astrocyte-derived Ang-1 and Shh alleviate TBI-induced BBB disruption. Therefore, astrocytes are key players in the regulation of BBB function, and astrocytic functional molecules may be attractive targets for the discovery of novel therapeutic drugs against TBI.
4. CANDIDATE FUNCTIONAL MOLECULES IN ASTROCYTES FOR DISCOVERING NOVEL THERAPEUTIC DRUGS FOR TBI
4.1. Endothelin ETB ReceptorEndothelins (ETs), including ET-1, -2, and -3, were initially discovered as vasoconstrictor peptides responsible for various pathophysiological responses including ischemia, inflammation, and BBB disruption in the damaged brain.35,36) ET receptors are seven-transmembrane G protein-coupled receptors and are classified into two distinct types: ETA and ETB receptors. In the injured mouse cerebrum after FPI, expression of the ETB receptor, but not the ETA receptor, is predominantly observed in GFAP-positive reactive astrocytes (Fig. 5A). Repeated i.c.v. administration of BQ788, a selective ETB receptor antagonist (Fig. 5B), decreases the number of GFAP-positive reactive astrocytes after FPI.1) As shown in Fig. 5C, BQ788 reduces the extravasation of Evans blue and FITC-dextran in the injured cerebrum after FPI. The FPI-induced increase in brain water content can also be reduced by BQ788.1) Furthermore, BQ788 decreases the expression of astrocytic MMP-9 and VEGF-A in the cerebrum after FPI, whereas it increases the expression of astrocytic Ang-1 and Shh.1,16,26)
Bosentan is a nonselective ET receptor antagonist used to treat pulmonary arterial hypertension. In the TBI mouse model, repeated intravenous (i.v.) administration of bosentan (Fig. 5B) reduces Evans blue extravasation into the injured cerebrum (Fig. 5D). The FPI-induced increase in brain water content is also reduced by bosentan.37) Furthermore, bosentan decreases the expression of ET-1, MMP-9, and VEGF-A in the injured cerebrum after FPI.37) In contrast, repeated i.v. administration of ambrisentan, a selective ETA antagonist (Fig. 5B), does not reduce FPI-induced Evans blue extravasation (Fig. 5D). Additionally, ambrisentan does not reduce brain water content in the injured cerebrum after FPI.37)
These results suggest that ETB receptor antagonists mitigate TBI-induced BBB disruption and brain edema by decreasing astrocytic vascular permeability-accelerating factors and increasing astrocytic vascular protective factors. Therefore, astrocytic ETB receptors may be attractive targets for developing therapeutic drugs for TBI.
4.2. Histamine H2 ReceptorHistamine is a major neurotransmitter that plays an essential role in multiple physiological reactions, including the sleep-wake cycle, learning, and memory in the central nervous system. Histamine receptors are classified into four types: H1, H2, H3, and H4. Some studies have suggested a relationship between H2 receptors and brain damage. H2 receptor antagonists reduced brain edema in experimental cerebral ischemia animal models.38) However, other studies have shown that H2 receptor antagonists aggravate ischemic damage in experimental cerebral ischemia model rats.39,40) Furthermore, H2 receptor agonists improve neuronal dysfunction by promoting astrocyte migration in cerebral ischemia rats.41,42) These findings imply that H2 receptors contribute to the pathological conditions of brain damage and regulate astrocyte functions. Mohanty et al.43) found that the increased brain water content as well as brain histamine levels were prevented by treatment with the histamine H2 receptor antagonist cimetidine in TBI model rats. Therefore, H2 receptors might be involved in TBI-induced pathological conditions.
In the injured mouse cerebrum after FPI, the expression level of the H2 receptor is increased,44) and GFAP-positive reactive astrocytes express H2 receptors (Fig. 6A). Repeated i.c.v. administration of selective H2 receptor antagonist ranitidine did not affect to Evans blue extravasation,44) whereas repeated i.c.v. administration of selective H2 receptor agonists, such as amthamine and dimaprit (Fig. 6B), reduce Evans blue extravasation in the injured cerebrum after FPI (Fig. 6C). A decrease in endothelial tight junction proteins causes BBB disruption following brain edema formation (Fig. 6D). Expression levels of tight junction proteins, such as claudin-5 and occludin, decrease in the injured cerebrum after FPI, whereas amthamine and dimaprit increase their expression levels (Fig. 6E). Furthermore, amthamine and dimaprit increase the expression levels of vascular protective factors, such as Ang-1 and Shh.44) In cultured astrocytes, treatment with amthamine or dimaprit increases the expression of Ang-1 and Shh.44) These results suggest that H2 receptor agonists reduce TBI-induced BBB disruption by increasing tight junction proteins and astrocyte-derived vascular protective factors. Therefore, the astrocytic H2 receptor might also be a target functional molecule for the development of therapeutic drugs for TBI.
4.3. Transient Receptor Potential Vanilloid 4 (TRPV4)TRPV4 is a calcium-permeable, nonselective cation channel sensitive to temperature and osmotic pressure. TRPV4 contributes to various physiological conditions, including the regulation of neuronal excitability and proliferation of brain cells,45,46) and is responsible for various pathological conditions of TBI, including neuronal death, inflammation, and brain edema.47–50) Lu et al.48) suggested that administration of the TRPV4 antagonist RN1734 significantly attenuated TBI-induced brain edema through decreasing the phosphorylation of mitogen-activated protein/extracellular signal-regulated kinase kinase (MEK), extracellular signal-regulated kinase (ERK), and protein kinase B (Akt) proteins.
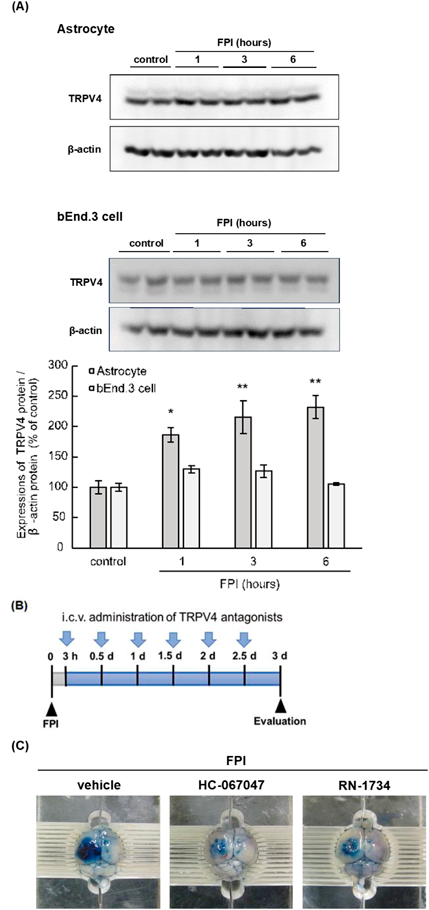
We previously found that astrocytes expressed TRPV4, and that the expression level of astrocytic TRPV4 was increased by FPI (Fig. 7A). In contrast, FPI did not increase TRPV4 expression in brain endothelial cells (bEnd.3 cells) (Fig. 7A). Repeated i.c.v. administration of selective TRPV4 antagonists, such as HC-067047 and RN-1734 (Fig. 7B), reduced Evans blue extravasation in the injured mouse cerebrum after FPI (Fig. 7C). HC-067047 and RN-1734 also reduced FPI-induced increases in brain water content.34) Furthermore, HC-067047 and RN-1734 decreased expression of ET-1, MMP-9, and VEGF-A in the injured cerebrum after FPI.34) In cultured astrocytes, treatment with HC-067047 or RN-1734 decreased expression of ET-1, MMP-9, and VEGF-A after FPI.34) These results suggest that TRPV4 antagonists alleviate TBI-induced BBB disruption and brain edema by decreasing the levels of astrocyte-derived vascular permeability-accelerating factors. Therefore, astrocytic TRPV4 may be a candidate functional molecule for the development of therapeutic drugs for TBI.
5. DISCUSSION
TBI-induced BBB disruption causes brain edema and neuroinflammation, resulting in unexpected death or serious disability. Therefore, the protection and recovery of the BBB may be a significant strategy for TBI. Since astrocyte-derived bioactive factors exert protective and detrimental actions against TBI-induced BBB disruption, the regulation of astrocyte function is essential for BBB protection. We have found that reactive astrocytes predominantly expressed endothelin ETB receptor, histamine H2 receptor, and TRPV4 in the injured cerebrum after TBI, and the administration of ETB receptor antagonists, H2 receptor agonists, and TRPV4 antagonists alleviated BBB disruption and brain edema in an experimental TBI model mice.1,34,44) Therefore, astrocytic functional molecules are important candidates for the development of therapeutic drugs against TBI.
Intravenous administration may be selected for introducing therapeutic drugs for TBI because emergent treatments are essential for TBI-induced BBB disruption and brain edema. We found that intravenous administration of BQ788 mitigated TBI-induced BBB disruption and brain edema in experimental model mice.1) However, delivery of BQ788 from the peripheral tissue into the central nervous system may be limited owing to its peptide-derived properties, which may require high doses of BQ788 for clinical use. Therefore, the development of alternative ETB receptor antagonists to facilitate brain transition may be essential in the future. Bosentan is a candidate therapeutic drug for treating TBI. Bosentan has already been used clinically as a therapeutic drug for pulmonary arterial hypertension, and we found that i.v. administration of bosentan alleviated BBB disruption and brain edema in experimental TBI model mice.37) Therefore, drug repositioning of bosentan for TBI is expected to occur in the future.
We showed that i.v. administration of H2 receptor agonists alleviate TBI-induced BBB disruption and increase expression levels of vascular protective factors in experimental mice.44) In contrast, intravenous administration of H2 receptor agonists decreases blood pressure in both sham and TBI model mice.44) Therefore, blood pressure management may be essential during the administration of H2 receptor agonists. Furthermore, H2 receptor agonists may only be administered for several days in the acute phase of TBI because persistent administration may cause gastrointestinal tract disturbances by accelerating gastric acid secretion. When these problems are resolved, H2 receptor agonists can be used as therapeutic drugs to treat TBI-induced BBB disruption.
We also found that i.c.v. administration of TRPV4 antagonists alleviates BBB disruption and brain edema in an experimental TBI model mice.34) However, we did not investigate the effects of i.v. administration on TBI-induced BBB disruption and brain edema. Liao et al.51) showed that intravenous administration of HC-067047 prevents shockwave-induced BBB disruption in experimental rats and that intravenous administration of TRPV4 antagonists might reduce TBI-induced BBB disruption, which should be investigated in the future.
Our studies suggest that astrocyte-targeting drugs can alleviate TBI-induced BBB disruption and brain edema by regulating the expression of astrocyte-derived bioactive factors (Fig. 8). Although appropriate administration routes, doses, side effects, and clinical trials should be investigated in the future, drugs targeting functional molecules in astrocytes are expected to become novel therapeutics for TBI.
Acknowledgments
I would like to acknowledge Dr. Yutaka Koyama of Kobe Pharmaceutical University, Dr. Shigeru Hishinuma of Meiji Pharmaceutical University, Dr. Hiroyuki Mizuguchi of Osaka-Ohtani University, Dr. Tomokazu Watano of Osaka-Ohtani University, Dr. Yasuhiro Ogawa of Meiji Pharmaceutical University, and Dr. Kahori Shimizu of Osaka-Ohtani University for their assistance. This work was supported by a Grant-in-Aid for Young Scientists (Grant Number: 20K16016) from the Japan Society for the Promotion of Science.
Conflict of Interest
The author declares no conflict of interest.
Notes
This review of the author’s work was written by the author upon receiving the 2023 Pharmaceutical Society of Japan Award for Young Scientists.
REFERENCES
- 1) Michinaga S, Kimura A, Hatanaka S, Minami S, Asano A, Ikushima Y, Matsui S, Toriyama Y, Fujii M, Koyama Y. Delayed administration of BQ788, an ETB antagonist, after experimental traumatic brain injury promotes recovery of blood-brain barrier function and a reduction of cerebral edema in mice. J. Neurotrauma, 35, 1481–1494 (2018).
- 2) Yang DX, Jing Y, Liu YL, Xu ZM, Yuan F, Wang ML, Geng Z, Tian HL. Inhibition of transient receptor potential vanilloid 1 attenuates blood-brain barrier disruption after traumatic brain injury. in mice. J. Neurotrauma, 36, 1279–1290 (2019).
- 3) Thal SC, Neuhaus W. The blood-brain barrier as a target in traumatic brain injury treatment. Arch. Med. Res., 45, 698–710 (2014).
- 4) Alluri H, Wiggins-Dohlvik K, Davis ML, Huang JH, Tharakan B. Blood–brain barrier dysfunction following traumatic brain injury. Metab. Brain Dis., 30, 1093–1104 (2015).
- 5) Michinaga S, Koyama Y. Protection of the blood–brain barrier as a therapeutic strategy for brain damage. Biol. Pharm. Bull., 40, 569–575 (2017).
- 6) Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience, 129, 1021–1029 (2004).
- 7) Jha RM, Kochanek PM, Simard JM. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology, 145 (Pt. B), 230–246 (2019).
- 8) Faden AI, Wu J, Stoica BA, Loane DJ. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br. J. Pharmacol., 173, 681–691 (2016).
- 9) Sulhan S, Lyon KA, Shapiro LA, Huang JH. Neuroinflammation and blood–brain barrier disruption following traumatic brain injury: pathophysiology and potential therapeutic targets. J. Neurosci. Res., 98, 19–28 (2020).
- 10) Michinaga S, Koyama Y. Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage. Int. J. Mol. Sci., 20, 571 (2019).
- 11) Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci., 28, 7231–7243 (2008).
- 12) Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br. J. Pharmacol., 173, 692–702 (2016).
- 13) Castejón OJ. Morphological astrocytic changes in complicated human brain trauma. A light and electron microscopic study. Brain Inj., 12, 409–427, discussion, 407 (1998).
- 14) Dunn C, Sturdivant N, Venier S, Ali S, Wolchok J, Balachandran K. Blood–brain barrier breakdown and astrocyte reactivity evident in the absence of behavioral changes after repeated traumatic brain injury. Neurotrauma Rep., 2, 399–410 (2021).
- 15) Zhai Y, Ye SY, Wang QS, Xiong RP, Fu SY, Du H, Xu YW, Peng Y, Huang ZZ, Yang N, Zhao Y, Ning YL, Li P, Zhou YG. Overexpressed ski efficiently promotes neurorestoration, increases neuronal regeneration, and reduces astrogliosis after traumatic brain injury. Gene Ther., 30, 75–87 (2023).
- 16) Michinaga S, Tanabe A, Nakaya R, Fukutome C, Inoue A, Iwane A, Minato Y, Tujiuchi Y, Miyake D, Mizuguchi H, Koyama Y. Angiopoietin-1/Tie-2 signal after focal traumatic brain injury is potentiated by BQ788, an ETB receptor antagonist, in the mouse cerebrum: involvement in recovery of blood-brain barrier function. J. Neurochem., 154, 330–348 (2020).
- 17) Lo AC, Chen AY, Hung VK, Yaw LP, Fung MK, Ho MC, Tsang MC, Chung SS, Chung SK. Endothelin-1 overexpression leads to further water accumulation and brain edema after middle cerebral artery occlusion via aquaporin 4 expression in astrocytic end-feet. J. Cereb. Blood Flow Metab., 25, 998–1011 (2005).
- 18) Gao W, Zhao Z, Yu G, Zhou Z, Zhou Y, Hu T, Jiang R, Zhang J. VEGI attenuates the inflammatory injury and disruption of blood-brain barrier partly by suppressing the TLR4/NF-κB signaling pathway in experimental traumatic brain injury. Brain Res., 1622, 230–239 (2015).
- 19) Min H, Hong J, Cho IH, Jang YH, Lee H, Kim D, Yu SW, Lee S, Lee SJ. TLR2-induced astrocyte MMP9 activation compromises the blood brain barrier and exacerbates intracerebral hemorrhage in animal models. Mol. Brain, 8, 23 (2015).
- 20) Michinaga S, Seno N, Fuka M, Yamamoto Y, Minami S, Kimura A, Hatanaka S, Nagase M, Matsuyama E, Yamanaka D, Koyama Y. Improvement of cold injury-induced mouse brain edema by endothelin ETB antagonists is accompanied by decreases in matrixmetalloproteinase 9 and vascular endothelial growth factor-A. Eur. J. Neurosci., 42, 2356–2370 (2015).
- 21) Zhang ZG, Zhang L, Croll SD, Chopp M. Angiopoietin-1 reduces cerebral blood vessel leakage and ischemic lesion volume after focal cerebral embolic ischemia in mice. Neuroscience, 113, 683–687 (2002).
- 22) Yu H, Wang P, An P, Xue Y. Recombinant human angiopoietin-1 ameliorates the expressions of ZO-1, occludin, VE-cadherin, and PKCα signaling after focal cerebral ischemia/reperfusion in rats. J. Mol. Neurosci., 46, 236–247 (2012).
- 23) Xia YP, He QW, Li YN, Chen SC, Huang M, Wang Y, Gao Y, Huang Y, Wang MD, Mao L, Hu B. Recombinant human sonic hedgehog protein regulates the expression of ZO-1 and occludin by activating angiopoietin-1 in stroke damage. PLOS ONE, 8, e68891 (2013).
- 24) Chen SC, Huang M, He QW, Zhang Y, Opoku EN, Yang H, Jin HJ, Xia YP, Hu B. Administration of sonic hedgehog protein induces angiogenesis and has therapeutic effects after stroke in rats. Neuroscience, 352, 285–295 (2017).
- 25) Xing G, Zhao T, Zhang X, Li H, Li X, Cui P, Li M, Li D, Zhang N, Jiang W. Astrocytic sonic hedgehog alleviates intracerebral hemorrhagic brain injury via modulation of blood-brain barrier integrity. Front. Cell. Neurosci., 14, 575690 (2020).
- 26) Michinaga S, Inoue A, Sonoda K, Mizuguchi H, Koyama Y. Down-regulation of astrocytic sonic hedgehog by activation of endothelin ETB receptors: involvement in traumatic brain injury-induced disruption of blood brain barrier in a mouse model. Neurochem. Int., 146, 105042 (2021).
- 27) Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. U.S.A., 106, 1977–1982 (2009).
- 28) Yang Y, Rosenberg GA. MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol. Biol., 762, 333–345 (2011).
- 29) Boroujerdi A, Welser-Alves JV, Milner R. Matrix metalloproteinase-9 mediates post-hypoxic vascular pruning of cerebral blood vessels by degrading laminin and claudin-5. Angiogenesis, 18, 255–264 (2015).
- 30) Kim JY, Ko AR, Hyun HW, Kang TC. ETB receptor-mediated MMP-9 activation induces vasogenic edema via ZO-1 protein degradation following status epilepticus. Neuroscience, 304, 355–367 (2015).
- 31) Michinaga S. The endothelin ETB receptor antagonist BQ788 protects against brain edema after fluid percussion injury by decreasing vascular endothelial growth factor-A expression in mice. Yakugaku Zasshi, 137, 1241–1246 (2017).
- 32) Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T. A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J. Neurochem., 89, 503–513 (2004).
- 33) Alvarez JI, Dodelet-Devillers A, Kebir H, Ifergan I, Fabre PJ, Terouz S, Sabbagh M, Wosik K, Bourbonnière L, Bernard M, van Horssen J, de Vries HE, Charron F, Prat A. The Hedgehog pathway promotes blood–brain barrier integrity and CNS immune quiescence. Science, 334, 1727–1731 (2011).
- 34) Michinaga S, Onishi K, Shimizu K, Mizuguchi H, Hishinuma S. Pharmacological inhibition of transient receptor potential vanilloid 4 reduces vasogenic edema after traumatic brain injury in mice. Biol. Pharm. Bull., 44, 1759–1766 (2021).
- 35) Schinelli S. Pharmacology and physiopathology of the brain endothelin system: an overview. Curr. Med. Chem., 13, 627–638 (2006).
- 36) Dashwood MR, Loesch A. Endothelin-1 as a neuropeptide: neurotransmitter or neurovascular effects? J. Cell Commun. Signal., 4, 51–62 (2010).
- 37) Michinaga S, Inoue A, Yamamoto H, Ryu R, Inoue A, Mizuguchi H, Koyama Y. Endothelin receptor antagonists alleviate blood-brain barrier disruption and cerebral edema in a mouse model of traumatic brain injury: a comparison between bosentan and ambrisentan. Neuropharmacology, 175, 108182 (2020).
- 38) Tósaki A, Szerdahelyi P, Joó F. Treatment with ranitidine of ischemic brain edema. Eur. J. Pharmacol., 264, 455–458 (1994).
- 39) Adachi N, Seyfried FJ, Arai T. Blockade of central histaminergic H2 receptors aggravates ischemic neuronal damage in gerbil hippocampus. Crit. Care Med., 29, 1189–1194 (2001).
- 40) Otsuka R, Adachi N, Hamami G, Liu K, Yorozuya T, Arai T. Blockade of central histaminergic H2 receptors facilitates catecholaminergic metabolism and aggravates ischemic brain damage in the rat telencephalon. Brain Res., 974, 117–126 (2003).
- 41) Hamami G, Adachi N, Liu K, Arai T. Alleviation of ischemic neuronal damage by histamine H2 receptor stimulation in the rat striatum. Eur. J. Pharmacol., 484, 167–173 (2004).
- 42) Liao RJ, Jiang L, Wang RR, Zhao HW, Chen Y, Li Y, Wang L, Jie LY, Zhou YD, Zhang XN, Chen Z, Hu WW. Histidine provides long-term neuroprotection after cerebral ischemia through promoting astrocyte migration. Sci. Rep., 5, 15356 (2015).
- 43) Mohanty S, Dey PK, Sharma HS, Singh S, Chansouria JP, Olsson Y. Role of histamine in traumatic brain edema: an experimental study in the rat. J. Neurol. Sci., 90, 87–97 (1989).
- 44) Michinaga S, Sonoda K, Inazuki N, Ezaki M, Awane H, Shimizu K, Hishinuma S, Mizuguchi H. Selective histamine H2 receptor agonists alleviate blood-brain barrier disruption by promoting the expression of vascular protective factors following traumatic brain injury in mice. J. Pharmacol. Sci., 150, 135–145 (2022).
- 45) Hatano N, Suzuki H, Itoh Y, Muraki K. TRPV4 partially participates in proliferation of human brain capillary endothelial cells. Life Sci., 92, 317–324 (2013).
- 46) Men C, Wang Z, Zhou L, Qi M, An D, Xu W, Zhan Y, Chen L. Transient receptor potential vanilloid 4 is involved in the upregulation of connexin expression following pilocarpine-induced status epilepticus in mice. Brain Res. Bull., 152, 128–133 (2019).
- 47) Jie P, Hong Z, Tian Y, Li Y, Lin L, Zhou L, Du Y, Chen L, Chen L. Activation of transient receptor potential vanilloid 4 induces apoptosis in hippocampus through downregulating PI3K/Akt and upregulating p38 MAPK signaling pathways. Cell Death Dis., 6, e1775 (2015).
- 48) Lu KT, Huang TC, Tsai YH, Yang YL. Transient receptor potential vanilloid type 4 channels mediate Na-K-Cl-co-transporter-induced brain edema after traumatic brain injury. J. Neurochem., 140, 718–727 (2017).
- 49) Hoshi Y, Okabe K, Shibasaki K, Funatsu T, Matsuki N, Ikegaya Y, Koyama R. Ischemic brain injury leads to brain edema via hyperthermia-induced TRPV4 activation. J. Neurosci., 38, 5700–5709 (2018).
- 50) Wang Z, Zhou L, An D, Xu W, Wu C, Sha S, Li Y, Zhu Y, Chen A, Du Y, Chen L, Chen L. TRPV4-induced inflammatory response is involved in neuronal death in pilocarpine model of temporal lobe epilepsy in mice. Cell Death Dis., 10, 386 (2019).
- 51) Liao WH, Hsiao MY, Kung Y, Liu HL, Béra JC, Inserra C, Chen WS. TRPV4 promotes acoustic wave-mediated BBB opening via Ca2+/PKC-δ pathway. J. Adv. Res., 26, 15–28 (2020).