Abstract
Microglia-mediated neuroinflammation plays a critical role in the onset and progression of Alzheimer’s disease. In a previous study, we synthesized 6-hydroxy-3′-propyl-[1,1′-biphenyl]-3-propanoic acid (6OHA) based on the structure of magnaldehyde B, a natural compound that our group identified as a retinoid X receptor (RXR) agonist. However, its potential effects on inflammation in microglial cells remain unexplored. In this study, we specifically focused on the early-phase inflammatory responses to lipopolysaccharide (LPS) and evaluated the inhibitory effects of 6OHA on BV-2 microglial cells following 2 h of LPS exposure. Similar to the existing RXR agonist bexarotene (Bex), 6OHA treatment (0.1 and 1 μM) resulted in a dose-dependent decrease in the mRNA levels of proinflammatory mediators, including interleukin-1β (Il1b), Il6, and inducible nitric oxide synthase. However, these effects on proinflammatory mediators were effectively abolished by the RXR antagonist UVI3003. Additionally, 6OHA promoted M2 microglia polarization after 24 h of treatment, as evidenced by the increased mRNA levels of the M2 marker genes arginase-1 (Arg1), C-C motif chemokine ligand 6 (Ccl6), Ccl17, and Ccl22. Notably, 6OHA induced a distinct set of M2 microglial markers compared with IL-4, a known M2 microglial inducer. Furthermore, the transcription of Arg1, a key M2 marker gene, is regulated by retinoic acid receptor/RXR heterodimers and the IL-4 signaling pathway. Collectively, 6OHA suppressed the early inflammatory responses to LPS and promoted M2 microglial polarization through a mechanism distinct from that of IL-4. Therefore, RXR agonists, including 6OHA and Bex, may exhibit dual anti-inflammatory effects and serve as novel modulators of neuroinflammation.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by the presence of β-amyloid plaques and tau neurofibrillary tangles; however, its etiology and pathogenesis remain unclear. Recent genome-wide association studies of late-onset AD have identified single-nucleotide polymorphisms associated with the risk of AD.1,2) Importantly, the large proportion of identified genes that are enriched or selectively expressed in microglia strongly suggests that innate immunity and inflammation may be implicated in AD pathogenesis.3) Recent accumulating evidence suggests that microglia-mediated neuroinflammation plays a critical role in the initiation and progression of AD.4)
Microglia are specialized macrophages of the central nervous system (CNS), accounting for 5–12% of the total cell population in the brain.5) Initially characterized as the innate immune cell of the brain, microglia also engage in the pruning of dendrite processes, regulation of their microenvironment, production and release of growth and survival factors, promotion of apoptosis, and clearance of cell detritus.6–8) Notably, most of these functions are dependent on the activation states of microglia, which are conveniently categorized as resting (M0), proinflammatory or classically activated (M1), and anti-inflammatory or alternatively activated (M2), based on functions and morphology.9) Under normal physiological conditions, microglia are involved in resolving the pathology and mitigating inflammation for the benefit of the host. By contrast, microglia adopt a detrimental phenotype under sustained or excessive exposure to insults, such as danger-associated molecular patterns, pathogen-associated molecular patterns, and inflammatory cytokines, initiating a vicious cycle that induces cellular and tissue damage.10) This damage, in turn, exacerbates the release of inflammatory mediators, further amplifying the inflammatory response and ultimately leading to neuronal and tissue damage, chronic inflammation, and the onset and progression of AD.11,12) Although the continuum of intermediate phenotypes between M1 and M2 is important and reflects the dynamic nature of microglial activation, modulating the transition of microglia between these two phenotypes, which determine either initiation or resolution of neuroinflammation, is crucial for protecting brain tissue and maintaining CNS homeostasis. Considering the critical role of neuroinflammation in the progression of AD, targeting microglial activity has emerged as a potential therapeutic strategy. Two potential strategies to mitigate neuroinflammation during AD progression include suppressing the production of early inflammatory mediators by M1 microglia and enhancing both the abundance and functional capacity of M2 microglia to facilitate inflammation resolution and tissue repair.
Retinoid X receptor (RXR) is a member of the nuclear receptor (NR) superfamily of ligand-activated transcription factors and serves as an obligatory heterodimer partner for numerous type II NRs, including peroxisome proliferator-activated receptor (PPAR), liver X receptor, retinoic acid receptor (RAR), and several others.13) In the context of their functions, RXRs are critical and distinct members of the NR superfamily involved in regulating various physiological processes, including cellular metabolism and neuronal function.14) Additionally, various NR agonists, including RXR agonists, have been shown to exhibit diverse beneficial effects in murine models of AD. These include promoting amyloid β (Aβ) clearance, suppressing Aβ generation, regulating neuronal function, and exerting anti-inflammatory effects.15)
With regard to inflammation in particular, the synthetic RXR agonist bexarotene (Bex) has been shown to alleviate or improve pathological conditions across various disease models, including neuropathic pain induced by chronic constriction injury,16) subarachnoid hemorrhage,17) traumatic brain injury,18) and lipopolysaccharide (LPS)-induced depression.19) These effects are suggested to be partly mediated by reducing the production of proinflammatory cytokines. Identifying molecular targets and elucidating the mechanisms underlying the anti-inflammatory effects observed in these in vivo studies remain challenging, particularly due to the pleiotropic nature of the actions of RXR agonist. However, evidence suggests that the actions of Bex are mediated via the PPARγ/RXR heterodimer pathway. The PPARγ/RXR heterodimer is classified as a permissive heterodimer, which can be activated by either a PPARγ agonist or an RXR agonist and is synergistically activated in the presence of both agonists.20) Therefore, the effects of Bex in inflammatory animal models suggest that it acts solely as an RXR agonist or synergistically engages the PPARγ/RXR heterodimer only in the presence of PPARγ agonists to exhibit potent anti-inflammatory effects.
Although several studies on the anti-inflammatory activity of PPARγ agonists in microglia suggest that they can inhibit the production of inflammatory cytokines and mediators, the results remain inconclusive.21–24) Additionally, PPARγ plays a crucial role in promoting the polarization of microglia to the M2 phenotype by inhibiting inflammatory transcription factors.25–27) Collectively, these findings suggest that endogenous or co-administered PPARγ agonists may enhance the neuroinflammation-suppressing effects of orally administered Bex in vivo. However, considering that thiazolidinedione-based PPARγ agonists, such as pioglitazone and rosiglitazone, exhibit minimal brain penetration due to their limited ability to cross the blood-brain barrier,28) and that PPARγ agonists exert their anti-inflammatory effects in vitro only at relatively high concentrations (>1–10 μM),24) it is likely that the effects of Bex are independent of PPARγ activity.
A few studies have shown that RXR agonists, including Bex, can suppress the production of inflammatory mediators in vitro, particularly in microglial cells.29,30) For example, in synovial cells, Bex exerts anti-inflammatory effects at concentrations of 150–300 nM, which are lower than the effective concentrations typically reported for PPARγ agonists.24,31) Similarly, 9-cis-retinoic acid (9-cis-RA) inhibits nitric oxide (NO) production and the expression of proinflammatory cytokines in microglial cells at a remarkably low concentration of 5 nM.32) However, 9-cis-RA has been shown to bind to both RXR and RAR with comparable affinity.33,34) This dual receptor agonist activity underscores the need for carefully interpreting the anti-inflammatory effects of 9-cis-RA, as RAR activation has been shown to attenuate neuroinflammation.35)
Although reports on the anti-inflammatory effects of RXR agonists in microglia are limited, their demonstrated neuroprotective properties,36–39) including the promotion of neurogenesis and enhancement of synaptic transmission—both of which are potentially critical for cognitive improvement—underscore the importance of a thorough investigation into their anti-inflammatory potential. Bex is currently used to treat cutaneous T-cell lymphoma40); however, its adverse effects, including hypertriglyceridemia, hypothyroidism, and hepatomegaly,41,42) limit its broader application to lifestyle-related diseases. Similarly, 9-cis-RA is associated with severe adverse effects, including headache and hypertriglyceridemia, and is further implicated in teratogenesis, limiting its potential application for AD.43,44)
We previously isolated naturally occurring RXR agonists and elaborated their biological activities.45–48) As part of our ongoing investigation of RXR agonists, we synthesized 6-hydroxy-3′-propyl-[1,1′-biphenyl]-3-propanoic acid (6OHA) based on the structure of magnaldehyde B, a natural product isolated from Magnolia obovata through RXR agonist screening.49,50) In this study, we investigated the effects of 6OHA on the induction and resolution of inflammation in microglial cells to advance our understanding of the biological activity of RXR agonists and to evaluate its potential as a therapeutic candidate for AD.
MATERIALS AND METHODS
Reagents
Bex was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan); UVI3003 was purchased from Cayman Chemical (Ann Arbor, MI, U.S.A.); Wy14643 and Am80 were obtained from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan); GW501516 was obtained from TargetMol Chemicals Inc. (Boston, MA, U.S.A.); Rosiglitazone was obtained from Alexia Biochemicals (San Diego, CA, U.S.A.); LPS (E.coli, O55:B5) was obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.); Mouse interleukin-4 (IL-4) was purchased from PeproTech (Cranbury, NJ, U.S.A.); phosphoric acid was obtained from Kishida Chemical (Osaka, Japan); and N-1-naphthylethylenediamine dihydrochloride, sulfanilamide, and sodium nitrite were purchased from Kanto Chemical (Tokyo, Japan). All chemicals were dissolved in dimethyl sulfoxide and stored at −20°C until use. Dulbecco’s modified Eagle’s medium (DMEM) and minimum essential medium (MEM) were obtained from FUJIFILM Wako Pure Chemical Corporation. Fetal bovine serum (FBS) was purchased from Biowest (Nuailé, France). Penicillin and streptomycin were obtained from FUJIFILM Wako Pure Chemical Corporation Corp.
Cell Culture
BV-2 cell line was obtained from Elabscience Bionovation Inc. (Houston, TX, U.S.A.). Human embryonic kidney 293 (HEK293) cells were provided by the RIKEN BioResource Center (Tsukuba, Japan) through the National Bio-Resource Project of the MEXT/AMED, Japan. BV-2 and HEK293 cells were maintained in MEM containing 10% FBS and non-essential amino acids (Sigma-Aldrich) at 37°C in a humidified atmosphere containing 5% CO2. MG5 microglial and A1 astrocyte-like cell lines (IFO50520 and IFO50519, respectively) were purchased from the Japanese Collection of Research Bioresource Cell Bank (Tokyo, Japan). We used MG5 cells derived from p53-deficient mice, due to their morphological, biochemical, and physiological similarities to primary-cultured microglia.51) MG5 cells were cultured in a mixture of 30% DMEM and 70% A1 cell-conditioned medium. Both media were supplemented with 10% FBS, 50 U/mL penicillin, and 50 μg/mL streptomycin at 37 °C in a humidified atmosphere containing 5% CO2.
Luciferase Reporter Assay
A luciferase reporter assay was performed according to our previously reported method.45) HEK293 cells were transfected via calcium phosphate co-precipitation with the following vectors: pCMX-hRXR-α and CRBPII-tk-Luc for the RXR reporter assay, along with the pCMX-β-gal expression vector and carrier DNA pUC18. Six hours after transfection, the cells were thoroughly washed with fresh medium and incubated with compounds at the indicated concentrations in a 10% FBS-containing medium for 40 h. Luciferase and β-galactosidase activities of the cell lysates were analyzed using a luminescence reader and spectrophotometer, respectively. Luciferase activity was normalized to the activity of an internal β-galactosidase control and expressed as the relative luciferase activity. Data are presented as the mean ± standard deviation (S.D.) of three determinants.
RT-Quantitative PCR (qPCR)
Total RNA was extracted using RNAiso Plus (TaKaRa Bio Inc., Kusatsu, Japan) according to the manufacturer’s instructions. RNA (300 ng) was reverse-transcribed using the ReverTra Ace qPCR RT Master Mix (TOYOBO Co., Ltd., Osaka, Japan). RT-qPCR was performed using the THUNDERBIRD SYBR qPCR Mix (TOYOBO Co., Ltd.) with the AB Step One Plus Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, U.S.A.). PCR was performed at the following conditions: 40 cycles of 3 s at 95°C and 30 s at 60°C. Relative mRNA expression was assessed using the ΔΔCt method, with β-actin as the endogenous control to normalize sample levels. All primer sequences used in this study are listed in Table 1.
Table 1. List of Primer Sequences Used for RT-qPCR
| Gene |
Forward Primer (5'→3') |
Reverse Primer (5'→3') |
| Tnfa |
CAGGCGGTGCCTATGTCTC |
CGATCACCCCGAAGTTCAGTAG |
| Il1b |
GTGTGGATCCCAAGCAATAC |
TTGTGAGGTGCTGATGTACC |
| Il6 |
CACGGCCTTCCCTACTTCA |
CATTTCCACGATTTCCCAGA |
| Nos2 |
AGTCACCAAAATGGCTCCCC |
GTGCAGCTTGTCCAGGGATT |
| Arg1 |
AAGAGTCAGTGTGGTGCTGG |
TGGTTGTCAGGGGAGTGTTG |
| Fizz1 |
GCTGGGATGACTGCTACTGG |
ACGAGTAAGCACAGGCAGTT |
| Cd206 |
CCTGGCAAGTATCCACAGCA |
CACTGGGGTTCCATCACTCC |
| Pparg |
CTCCAAGAATACCAAAGTGCGA |
GCCTGATGCTTTATCCCCACA |
| Ccl6 |
TGGGGCTTTGGAATGTGTCT |
CTCTATTGTGGCAGGGCGAA |
| Ccl17 |
CTCAGTGGAGTGTTCCAGGG |
GGCCTTGGGTTTTTCACCAA |
| Ccl22 |
CTACATCCGTCACCCTCTGC |
CTTCTTCACCCAGACCTGCC |
| Ccl24 |
GGCAGGGGTCATCTTCATCA |
CTGCCTTGGCCCCTTTAGAA |
| Actb |
CATCCGTAAAGACCTCTATGCCAA |
ATGGAGCCACCGATCCACA |
Cell culture supernatant was collected and centrifuged at 500 × g for 3 min and the resulting supernatant was used for ELISA. ELISA was performed using mouse IL-6 ELISA kit (R&D Systems, Minneapolis, MN, U.S.A.), mouse CCL6 ELISA kit, and Eotaxin-2 (CCL24) ELISA kit (RayBiotech, Peachtree Corners, GA, U.S.A.), according to the manufacturer’s instructions.
Measurement of NO Production
The nitrate levels in BV-2 cell culture supernatants were measured using Griess reagent. BV-2 cells were pretreated with 6OHA or Bex for 30 min, followed by incubation with 100 ng/mL LPS for 18 h. The culture supernatant was collected and mixed with Griess reagent [1 : 1 mixture (v/v) of 1% sulfanilamide and 0.1% N-1-naphthylethylenediamine dihydrochloride in 2.5% phosphoric acid] at the ratio of 100:100 μL in 96-well plates. After 20 min, the absorbance was measured at 550 nm using a microplate reader (Spark, Tecan, Switzerland).
Statistical Analyses
Data were analyzed using the GraphPad Prism 9.5.1 (GraphPad Software, San Diego, CA, U.SA.). Data are presented as the mean ± S.D. Statistical analyses were conducted using one-way ANOVA followed by Tukey’s post-hoc test or Dunnett’s test. A p-value <0.05 was considered statistically significant.
RESULTS
Inhibitory Effects of 6OHA on LPS-Induced Upregulation of Inflammatory Mediators in BV-2 Cells
RXRα luciferase reporter assay was performed to examine the RXR agonist activity of 6OHA, as previously reported.45) 6OHA was previously synthesized in our laboratory based on the structure of magnaldehyde B, a natural product with confirmed RXR agonist activity.49,50)
Figure 1 shows the chemical structures and RXR agonist activities of 6OHA and Bex. 6OHA exhibited potent RXRα agonist activity, with an EC50 value of 8.8 nM, comparable to that of the existing RXR agonist Bex (EC50: 18.7 nM).
To investigate the anti-inflammatory effect of the RXR agonist 6OHA, RT-qPCR was performed to examine time-dependent changes in the mRNA levels of tumor necrosis factor-α (Tnfa), interleukin-1β (Il1b), Il6, and inducible nitric oxide synthase (Nos2) following LPS stimulation. Tnfa mRNA levels increased as early as 0.5 h after LPS stimulation, followed by subsequent increases in Il1b, IL6, and Nos2 mRNA levels (Fig. 2). Therefore, we focused on the early responses to LPS stimulation and evaluated the inhibitory effects of 6OHA after 2 h of exposure. 6OHA treatment (0.1 and 1 μM) significantly suppressed the mRNA levels of Il1b, IL6, and Nos2, showing effects similar to those of Bex (Fig. 3). However, LPS-induced upregulation of Tnfa mRNA levels was not significantly suppressed by 6OHA or Bex. To investigate the inhibitory effect of 6OHA on the inflammatory mediators, the cells were treated with the RXR antagonist UVI3003. UVI3003 treatment effectively abolished the inhibitory effects of 6OHA on Il1b, IL6, and Nos2 mRNA levels (Fig. 4), indicating that the inhibitory effect of 6OHA was RXR-dependent.
Additionally, IL-1β and IL-6 protein levels and NO production were assessed using ELISA and NO assay, respectively. 6OHA and Bex treatments significantly suppressed LPS-induced upregulation of IL-6 and NO productions (Fig. 5). However, as previously reported in BV-2 cells,52) IL-1β was undetectable by ELISA following stimulation with a low concentration of LPS (100 ng/mL) for 18 h.
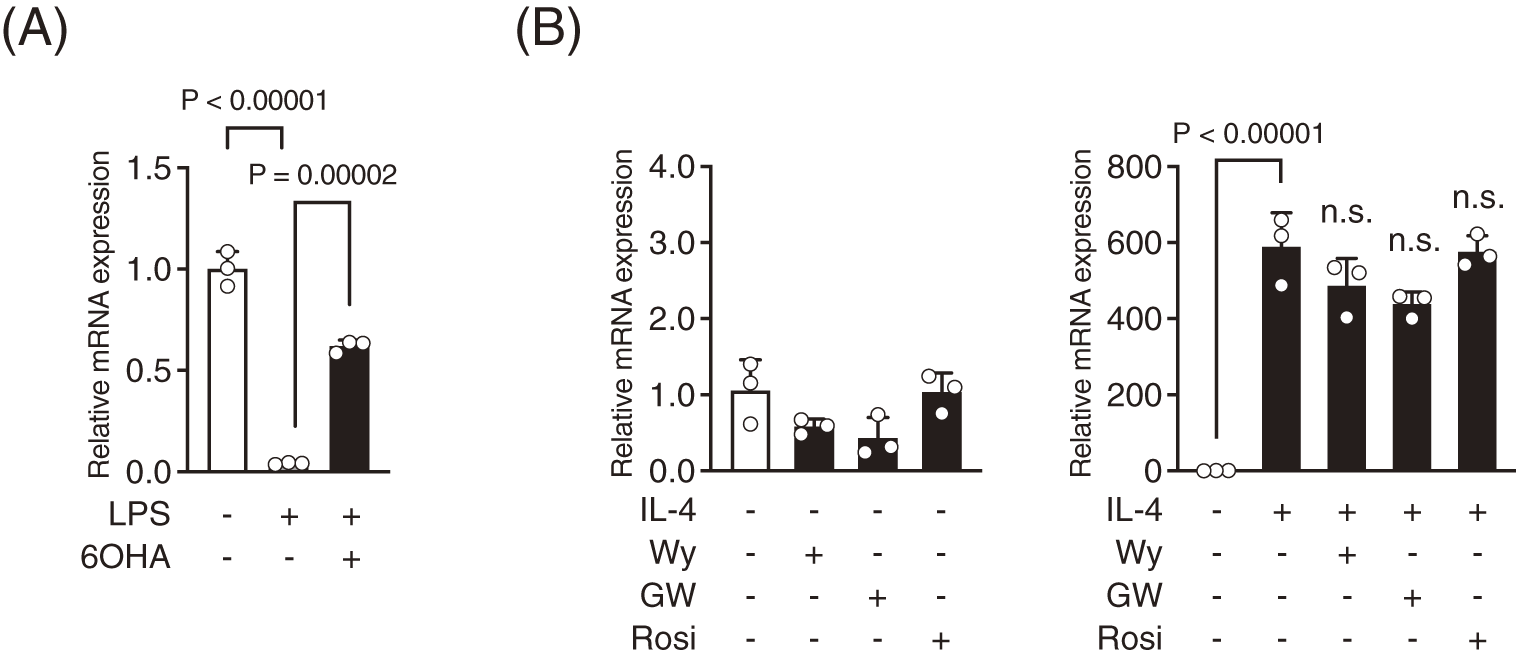
Treatment with relatively high concentrations (>50 μM) of PPARα and PPARγ agonists for an extended duration of 24 h has been shown to inhibit the production of proinflammatory cytokines in LPS-stimulated microglia.24,31) Therefore, we examined the effects of PPAR agonists on LPS-induced responses, focusing on the critical early phase within the first 2 h. Treatment with the PPARα agonist Wy14643, the PPARδ agonist GW501516, and the PPARγ agonist rosiglitazone did not suppress LPS-induced upregulation of Tnfa, Il1b, and Il6 mRNA levels at concentrations up to 10 μM (Supplementary Fig. 1).
Effects of 6OHA on Microglial Polarization to the M2 Phenotype
To investigate whether the RXR agonist 6OHA influences M2 microglial polarization, we examined the mRNA expression of M2-specific marker genes after 24-h treatment with 6OHA. To minimize the influence of cell-specific responses and obtain a generalized microglial reaction in M2 marker gene expression, we utilized two microglial cell lines: BV-2 and MG5. IL-4, a well-known inducer of M2 microglia polarization, increased the mRNA levels of typical M2 microglial marker genes, including arginase 1 (Arg1), resistin like-α (Fizz1), mannose receptor C type1 (Cd206), Pparg, and C-C moeps chemokine ligand 24 (Ccl24). By contrast, 6OHA treatment upregulated the mRNA expression levels of Arg1, Ccl6 (the mouse homolog of Ccl23), Ccl17, and Ccl22 (Fig. 6). Interestingly, Bex induced the mRNA expression levels of M2 microglial marker genes that were nearly identical to those induced by 6OHA, except for the notably higher expression levels of Fizz1 and Ccl24 mRNA in BV-2 cells (Supplementary Fig. 2). However, the mRNA expression of Fizz1 and Ccl17 was not detected in MG5 and BV-2 cells, respectively, following either treatment. Collectively, these results indicate that 6OHA and Bex may promote microglial polarization to the M2 phenotype, although they induce a distinct set of M2 microglial marker compared with IL-4. Additionally, there was a strong correlation between the mRNA and protein expression levels of CCL6 and CCL24 (Fig. 7), despite our inability to measure CCL6 in BV-2 cell culture media.
Moreover, LPS stimulation for 24 h significantly downregulated the mRNA levels of Arg1. Notably, 6OHA treatment attenuated the LPS-induced reduction in Arg1 mRNA levels in BV-2 cells (Fig. 8A).
Regarding the involvement of NRs in microglial polarization, research evidence suggests that PPARγ plays a critical role in microglial transition from the M1 to M2 phenotype and that the ability of IL-4 to induce M2 microglia is partly mediated through the activation of PPARγ.53) Considering that IL-4 significantly increased Pparg mRNA levels in BV-2 and MG5 cells (Fig. 6), we investigated whether PPARγ agonists can induce Arg1 mRNA expression in the presence or absence of IL-4. Treatment with PPARγ agonist rosiglitazone, as well as PPARα agonist and PPARδ agonist did not affect Arg1 mRNA expression, regardless of the presence or absence of IL-4 (Fig. 8B).
Furthermore, IL-4 and all-trans retinoic acid (RA) have been reported to synergistically upregulate Arg1 gene expression in RAW264.7 cells and mouse peritoneal macrophages.54) Therefore, we examined the effects of IL-4, 6OHA, and the RAR agonist Am80, individually or in combination, on Arg1 mRNA levels in BV-2 cells. Treatment with Am80 and 6OHA increased Arg1 mRNA expression, which was further enhanced following Am80 and 6OHA co-treatment (Fig. 9). Additionally, Am80 and 6OHA, either alone or in combination, synergistically increased Arg1 mRNA levels in the presence of IL-4. Collectively, these results suggest that the transcription of Arg1 is regulated in an interdependent manner by RAR/RXR heterodimers and IL-4 signaling pathways in microglia, but not by ligand-dependent transcriptional activation of PPARγ/RXR heterodimers.
Our study demonstrates that 6OHA and Bex exert inhibitory effects on the production of inflammatory mediators at early time points and promotes a shift to M2 microglial polarization.
DISCUSSION
Recent findings indicate that microglia are key regulators of neuroinflammation.3) Considering the strong association between neuroinflammation and the induction and progression of AD, strategies to mitigate inflammation––either by blocking its initiation or promoting its resolution––offer promising potential for AD prevention. In this study, we investigated the anti-inflammatory effects of the RXR agonist 6OHA with Bex as a positive control of a well-known RXR agonist, focusing specifically on its actions in microglia.
LPS stimulation upregulated the mRNA expression of proinflammatory cytokines and inflammatory mediators in BV-2 cells within 0.5–3 h (Fig. 2). To date, the anti-inflammatory effects of NR agonists in microglial cells have primarily been evaluated after extended LPS exposure, typically ranging from 6 to 24 h.24,31,55,56) Considering that LPS stimulation induces various pro-inflammatory cytokines, such as TNFα, IL-1β, and IL-6, which activate signaling pathways simultaneously, assessing the effects of NR agonists at later time points may obscure their direct mechanism of action. Therefore, we focused on the early responses to LPS and evaluated the effects of 6OHA on mRNA expression levels 2 h after LPS stimulation.
Similar to Bex, 6OHA treatment (0.1 and 1 μM) significantly downregulated the mRNA levels of Il1b, IL6, and Nos2 in a dose-dependent manner (Fig. 3). However, these effects were effectively abolished by the RXR antagonist UVI3003 (Fig. 4), indicating that the effects of 6OHA were RXR-dependent. Notably, 6OHA and Bex did not affect the mRNA levels of Tnfa (Fig. 3), suggesting that the early effects of RXR agonists could be gene-specific, similar to PPARγ or LXR agonists, which transrepress the transcription of inflammatory response genes in a gene-specific manner through the conjugation of Small Ubiquitin-like Modifier to PPARγ or LXR.57)
To investigate the mechanism by which 6OHA downregulates the mRNA levels of inflammatory mediators during the early response, we examined the effects of agonists targeting PPAR subtypes that inhibit cytokine production in microglial cells.24,31) Importantly, none of the agonists for PPAR subtypes inhibited the mRNA expression levels of proinflammatory cytokines at a concentration of 10 μM, whereas 6OHA exhibited significant inhibitory effects at 1 μM (Supplementary Fig. 1). Although previous studies have reported the anti-inflammatory effects of PPAR agonists,24,31) these effects were observed only at concentrations exceeding 50 μM and after prolonged incubation for 24 h. Overall, our findings suggest that the inhibitory effect of 6OHA on early responses to LPS may be independent of PPAR/RXR heterodimer activation.
Additionally, PPARγ agonists have been shown to exert anti-inflammatory effects by inhibiting activation pathways of transcription factors such as NF-κB and AP-1.21,23) By contrast, our preliminary findings indicate that 6OHA did not affect the nuclear translocation of NF-κB following LPS stimulation, suggesting that the suppressive effect of 6OHA on the early response to LPS may be independent of NF-κB. However, further investigation is required to elucidate the underlying mechanism, particularly whether 6OHA exerts its anti-inflammatory effects through RXR homodimers, RXR heterodimers with other NRs, or RXR monomers.
Furthermore, we investigated whether 6OHA can promote the polarization of microglial BV-2 and MG5 cells to the M2 phenotype. IL-4, which is known to facilitate the polarization of microglia from the M1 to the M2 phenotype,58) significantly upregulated the mRNA expression of typical M2 microglial markers, including Arg1, Fizz1, Cd206, Pparg, and Ccl24, in BV-2 and MG5 cells (Fig. 6). 6OHA also upregulated the mRNA expression of Arg1, Ccl6, Ccl17, and Ccl22, but did not increase Cd206, Pparg, and Ccl24 mRNA expression. Although Ccl6 is not a typical M2 microglial marker, it has been shown to promote M2 polarization and inhibit macrophage autophagy during skin wound healing, suggesting its involvement in inflammation resolution and tissue repair.59) M2 macrophage/microglia can be further classified into M2a, M2b, and M2c phenotypes. The M2a phenotype, induced by IL-4 or IL-13, plays an important role in wound healing and tissue repair by facilitating the removal of cellular debris and regeneration.60) In this study, the specific genes induced by IL-4 and 6OHA differ, but these genes are classified as M2a markers or are known to promote M2 polarization in macrophages and microglia.59,61) Similarly, Bex induced mRNA expression levels of M2 microglial marker genes that were nearly identical to those induced by 6OHA, although there are a few exceptions. Therefore, 6OHA and Bex appear to promote microglia polarization to the M2 phenotype (particularly M2a) in a manner that is similar to, yet distinct from IL-4. Collectively, these results suggest that RXR agonists, including 6OHA and Bex, may contribute to resolving inflammation and promoting tissue repair.
IL-4-induced polarization of microglia and macrophage to the M2 phenotype involves key transcription factors, including signal transducer and activator 6 (STAT6), interferon regulatory factor 4, and PPARγ.53) In this study, IL-4 induced Pparg mRNA expression (Fig. 6); however, the PPARγ agonist rosiglitazone failed to increase the expression of the M2 marker gene Arg1, either alone or in combination with IL-4 (Fig. 8B). Szanto et al. reported that IL-4 enhanced PPARγ expression and its ligand-induced transcriptional activity through an interaction between PPARγ and STAT6 at the promoters of PPARγ target genes in macrophages. However, ligand-dependent induction of Arg1 and Fizz1 mRNA levels was not observed.62) Overall, this result suggests that PPARγ is required to facilitate microglial polarization to the M2 phenotype. However, this effect may not be mediated through ligand-dependent transcriptional activation of PPARγ/RXR heterodimers, but rather through the regulation of specific genes by a transcription factor whose activity is influenced by changes in chromatin conformation upon ligand-independent binding of PPARγ/RXR heterodimers to specific chromatin regions.63) This suggests that RXR agonists do not promote microglial polarization to the M2 phenotype through the activation of PPARγ/RXR heterodimers.
A previous study showed that IL-4 and the RAR agonist RA synergistically upregulated Arg1 mRNA expression in mouse macrophages.54) In this study, 6OHA upregulated Arg1 mRNA expression alone or in combination with the RAR agonist Am80, which was further enhanced by IL-4 (Fig. 9). Arg1 is a prototypical M2 microglial marker that plays key roles in anti-inflammatory responses, immunosuppression, and wound healing by catalyzing the hydrolysis of arginine to produce ornithine, a substrate of polyamines and proline.64) By contrast, NOS2 is a prototypical M1 microglia marker that promotes inflammation through NO production. Therefore, the upregulation of Arg1 mRNA levels via RAR/RXR heterodimers or the IL-4 signaling pathway, combined with the suppression of Nos2 mRNA by RXR agonists, contributes to the reduction of NO synthesis by NOS2 and promotes microglial polarization to the M2 phenotype. Additionally, 6OHA significantly reversed LPS-induced downregulation of Arg1 mRNA levels in BV-2 cells (Fig. 8A). Although the data are not shown, the RAR agonist Am80 at 10 μM had no effect on the induction of Tnfa, Il1b, Il6, and Nos2 mRNA expression following LPS treatment, leading us to conclude that RAR/RXR heterodimers are not involved in an anti-inflammatory effect during the early phase of LPS-induced inflammation. By contrast, RAR/RXR heterodimers may play a critical role in promoting microglial polarization. Collectively, these findings suggest that 6OHA inhibits LPS-induced decrease in Arg1 mRNA levels and suppresses Nos2 mRNA levels, thereby mitigating inflammation and tissue damage. Furthermore, the similarities between 6OHA and Bex in their effects on inflammation (Figs. 3 and 5) and microglial polarization (Supplementary Fig. 2) suggest that these effects may represent common properties of RXR agonists. However, our unpublished data indicate that 6OHA does not induce hypertriglyceridemia or hypothyroidism, which are frequently observed adverse effects in patients with cutaneous T-cell lymphoma treated with Bex,41,42) suggesting that 6OHA may be a safer and more effective candidate for the treatment of inflammatory diseases or AD than Bex. Overall, this study advances our understanding of the biological activity and therapeutic potential of RXR agonists.
Taken together, RXR agonists, including 6OHA and Bex, may inhibit the production of inflammatory mediators in a gene-specific manner during the early responses to LPS and promote microglia polarization to the M2 phenotype through a mechanism similar to but distinct from that of IL-4, potentially accelerating the resolution of inflammation. To our knowledge, this is the first report demonstrating the dual ability of RXR agonists to modulate microglial inflammation, although the precise mechanisms require further investigation.
Acknowledgments
This work was supported by the JSPS KAKENHI (Grant No.: JP21K06636).
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Materials
This article contains supplementary materials.
REFERENCES
- 1) Jansen IE, Savage JE, Watanabe K, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet., 51, 404–413 (2019).
- 2) Wightman DP, Jansen IE, Savage JE, et al. A genome-wide association study with 1,126,653 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet., 53, 1276–1282 (2021).
- 3) Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener., 12, 43 (2017).
- 4) Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, Lamb BT. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. (N. Y.), 4, 575–590 (2018).
- 5) Lawson LJ, Perry VH, Dri P, Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience, 39, 151–170 (1990).
- 6) Spittau B. Aging microglia-phenotypes, functions and implications for age-related neurodegenerative diseases. Front. Aging Neurosci, 9, 194 (2017).
- 7) Bar E, Barak B. Microglia roles in synaptic plasticity and myelination in homeostatic conditions and neurodevelopmental disorders. Glia, 67, 2125–2141 (2019).
- 8) Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J. Cell Biol., 217, 459–472 (2018).
- 9) Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol., 11, 775–787 (2011).
- 10) Venegas C, Heneka MT. Danger-associated molecular patterns in Alzheimer’s disease. J. Leukoc. Biol., 101, 87–98 (2017).
- 11) Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell, 140, 918–934 (2010).
- 12) Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat. Rev. Neurol., 6, 193–201 (2010).
- 13) Moutinho M, Codocedo JF, Puntambekar SS, Landreth GE. Nuclear receptors as therapeutic targets for neurodegenerative diseases: lost in translation. Annu. Rev. Pharmacol. Toxicol., 59, 237–261 (2019).
- 14) Sharma S, Shen T, Chitranshi N, Gupta V, Basavarajappa D, Sarkar S, Mirzaei M, You Y, Krezel W, Graham SL, Gupta V. Correction to: Retinoid X receptor: cellular and biochemical roles of nuclear receptor with a focus on neuropathological involvement. Mol. Neurobiol., 59, 2051 (2022).
- 15) Moutinho M, Landreth GE. Therapeutic potential of nuclear receptor agonists in Alzheimer’s disease. J. Lipid Res., 58, 1937–1949 (2017).
- 16) Gui Y, Duan S, Xiao L, Tang J, Li A. Bexarotent attenuated chronic constriction injury-induced spinal neuroinflammation and neuropathic pain by targeting mitogen-activated protein kinase phosphatase-1. J. Pain., 21, 1149–1159 (2020).
- 17) Zuo Y, Huang L, Enkhjargal B, Xu W, Umut O, Travis ZD, Zhang G, Tang J, Liu F, Zhang JH. Activation of retinoid X receptor by bexarotene attenuates neuroinflammation via PPARγ/SIRT6/FoxO3a pathway after subarachnoid hemorrhage in rats. J. Neuroinflammation, 16, 47 (2019).
- 18) He J, Huang Y, Liu H, Sun X, Wu J, Zhang Z, Liu L, Zhou C, Jiang S, Huang Z, Zhong J, Guo Z, Jiang L, Cheng C. Bexarotene promotes microglia/macrophages-specific brain-derived neurotrophic factor expression and axon sprouting after traumatic brain injury. Exp. Neurol., 334, 113462 (2020).
- 19) Yuan C, Dai C, Li Z, Zheng L, Zhao M, Dong S. Bexarotene improve depression-like behaviour in mice by protecting against neuro-inflammation and synaptic damage. Neurochem. Res., 45, 1500–1509 (2020).
- 20) Mangelsdorf DJ, Evans EM. The RXR heterodimers and orphan receptors. Cell, 83, 841–850 (1995).
- 21) Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature, 437, 759–763 (2005).
- 22) Park EJ, Park SY, Joe EH, Jou I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1(SOCS1) and SOCS3 in glia. J. Biol. Chem., 278, 14747–14752 (2003).
- 23) Xing B, Xin T, Hunter RL, Bing G. Pioglitazone inhibition of lipopolysaccharide-induced nitric oxide synthase is associated with altered activity of p38 MAP kinase and PI3K/Akt. J. Neuroinflammation., 5, 4 (2008).
- 24) Storer PD, Xu J, Chavis J, Drew PD. Peroxisome proliferator-activated receptor-gamma agonists inhibit the activation of microglia and astrocytes: implications for multiple sclerosis. J. Neuroimmunol., 161, 113–122 (2005).
- 25) Castro LVG, Gonçalves-de-Albuquerque CF, Silva AR. Polarization of microglia and its therapeutic potential in sepsis. Int. J. Mol. Sci., 23, 4925 (2022).
- 26) Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, Chawla A. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature, 447, 1116–1120 (2007).
- 27) Croasdell A, Duffney PF, Kim N, Lacy SH, Sime PJ, Phipps RP. PPARγ and the innate immune system mediate the resolution of inflammation. PPAR Res., 2015, 549691 (2015).
- 28) Zhang M, Hu M, Montera MA, Westlund KN. Sustained relief of trigeminal neuropathic pain by a blood-brain barrier penetrable PPAR gamma agonist. Mol. Pain., 15, 1744806919884498 (2019).
- 29) Wang W, Nakashima K, Hirai T, Inoue M. Anti-inflammatory effects of naturally occurring retinoid X receptor agonists isolated from Sophora tonkinensis Gagnep. via retinoid X receptor/liver X receptor heterodimers. J. Nat. Med., 73, 419–430 (2019).
- 30) Li Y, Xing Q, Wei Y, Zhao L, Zhang P, Han X, Wang J. Activation of RXR by bexarotene inhibits inflammatory conditions in huma rheumatoid arthritis fibroblast-like synoviocytes. Int. J. Mol. Med., 44, 1963–1970 (2019).
- 31) Xu J, Storer PD, Chavis JA, Racke MK, Drew PD. Agonists for the peroxisome proliferator-activated receptor-α and the retinoid X receptor inhibit inflammatory responses of microglia. J. Neurosci. Res., 81, 403–411 (2005).
- 32) Xu J, Drew PD. 9-Cis-retinoic acid suppresses inflammatory responses of microglia and astrocytes. J. Neuroimmunol., 171, 135–144 (2006).
- 33) Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, Thaller C. 9-Cis retinoic acid is a high affinity ligand for the retinoid X receptor. Cell, 68, 397–406 (1992).
- 34) Levin AA, Sturzenbecker LJ, Kazmer S, Bosakowski T, Huselton C, Allenby G, Speck J, Kratzeisen C, Rosenberger M, Lovey A, Grippo JF. 9-Cis retinoic acid stereoisomer binds and activates the nuclear receptor RXR α. Nature, 355, 359–361 (1992).
- 35) Tian Y, Liu B, Li Y, Zhang Y, Shao J, Wu P, Xu C, Chen G, Shi H. Activation of RARα receptor attenuates neuroinflammation after SAH via promoting M1-to-M2 phenotypic polarization of microglia and regulating Mafb/Msr1/PI3K-Akt/NF-κB pathway. Front. Immunol., 13, 839796 (2022).
- 36) Mariani MM, Malm T, Lamb R, Jay TR, Neilson L, Casali B, Medarametla L, Landreth GE. Neuronally-directed effects of RXR activation in a mouse model of Alzheimer’s disease. Sci. Rep., 7, 42270 (2017).
- 37) Santos-Gil DF, Arboledá G, Sandoval-Hernández AG. Retinoid X receptor activation promotes re-myelination in a very old triple transgenic mouse model of Alzheimer’s disease. Neurosci. Lett., 750, 135764 (2021).
- 38) Muñoz-Cabrera JM, Sandoval-Hernández AG, Niño A, Báez T, Bustos-Rangel A, Cardona-Gómez GP, Múnera A, Arboleda G. Bexarotene therapy ameliorates behavioral deficits and induces functional and molecular changes in very-old triple transgenic mice model of Alzheimer ́s disease. PLOS ONE, 14, e0223578 (2019).
- 39) Mounier A, Georgiev D, Nam KN, Fitz NF, Castranio EL, Wolfe CM, Cronican AA, Schug J, Lefterov I, Koldamova R. Bexarotene-activated retinoid X receptors regulate neuronal differentiation and dendritic complexity. J. Neurosci., 35, 11862–11876 (2015).
- 40) Duvic M, Hymes K, Heald P, Breneman D, Martin AG, Myskowski P, Crowley C, Yocum RC. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. J. Clin. Oncol., 19, 2456–2471 (2001).
- 41) Duvic M, Martin AG, Kim Y, Olsen E, Wood GS, Crowley CA, Yocum RC. Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma. Arch. Dermatol., 137, 581–593 (2001).
- 42) Sherman SI, Gopal J, Haugen BR, Chiu AC, Whaley K, Nowlakha P, Duvic M. Central hypothyroidism associated with retinoid X receptor-selective ligands. N. Engl. J. Med., 340, 1075–1079 (1999).
- 43) van der Wees J, Schilthuis JG, Koster CH, Diesveld-Schipper H, Folkers GE, van der Saag PT, Dawson MI, Shudo K, van der Burg B, Durston AJ. Inhibition of retinoic acid receptor-mediated signalling alters positional identity in the develping hindbrain. Development, 125, 545–556 (1998).
- 44) Miller VA, Rigas JR, Benedetti FM, Verret AL, Tong WP, Kris MG, Gill GM, Loewen GR, Truglia JA, Ulm EH, Warrell RPJr. Initial clinical trial of the retinoid receptor pan agonist 9-cis retinoic acid. Clin. Cancer Res., 2, 471–475 (1996).
- 45) Kotani H, Tanabe H, Mizukami H, Makishima M, Inoue M. Identification of a naturally occurring rexinoid, honokiol, that activates the retinoid X receptor. J. Nat. Prod., 73, 1332–1336 (2010).
- 46) Inoue M, Tanabe H, Nakashima K, Ishida Y, Kotani H. Rexinoids isolated from Sophora tonkinensis with a gene expression profile distinct from the synthetic rexinoid bexarotene. J. Nat. Prod., 77, 1670–1677 (2014).
- 47) Nakashima K, Murakami T, Tanabe H, Inoue M. Identification of a naturally occurring retinoid X receptor agonist from Brazilian green propolis. Biochim. Biophys. Acta, Gen. Subj., 1840, 3034–3041 (2014).
- 48) Kotani H, Tanabe H, Mizukami H, Amagaya S, Inoue M. A naturally occurring rexinoid, honokiol, can serve as a regulator of various retinoid x receptor heterodimers. Biol. Pharm. Bull., 35, 1–9 (2012).
- 49) Nakashima K, Yamaguchi E, Noritake C, Mitsugi Y, Goto M, Hirai T, Abe N, Sakai E, Oyama M, Itoh A, Inoue M. Discovery and SAR of natural-product-inspired RXR agonists with heterodimer selectivity to PPARδ-RXR. ACS Chem. Biol., 15, 1526–1534 (2020).
- 50) Nakashima K, Okamura M, Matsumoto I, Kameda N, Tsuboi T, Yamaguchi E, Itoh A, Inoue M. Regulation of adipogenesis through retinoid X receptor and/or peroxisome proliferator-activated receptor by designed lignans based on natural products in 3T3-L1 cells. J. Nat. Med., 77, 315–326 (2023).
- 51) Ohsawa K, Imai Y, Nakajima K, Kohsaka S. Generation and characterization of a microglial cell line, MG5, derived from a p53-deficient mouse. Glia, 21, 285–298 (1997).
- 52) Watters JJ, Sommer JA, Pfeiffer ZA, Prabhu U, Guerra AN, Bertics PJ. A differential role for the mitogen-activated protein kinases in lipopolysaccharide signaling: the MEK/ERK pathway is not essential for nitric oxide and interleukin 1beta production. J. Biol. Chem., 277, 9077–9087 (2002).
- 53) Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature, 400, 378–382 (1999).
- 54) Lee B, Wu CY, Lin YW, Park SW, Wei LN. Synergistic activation of Arg1 gene by retinoic acid and IL-4 involves chromatin remodeling for transcription initiation and elongation coupling. Nucleic Acids Res., 44, 7568–7579 (2016).
- 55) Lee WJ, Ham SA, Yoo H, Hwang JS, Yoo T, Paek KS, Lim DS, Han SG, Lee CH, Hong K, Seo HG. Activation of PPARδ attenuates neurotoxicity by inhibiting lipopolysaccharide-triggered glutamate release in BV-2 microglial cells. J. Cell. Biochem., 119, 5609–5619 (2018).
- 56) Zhang-Gandhi CX, Drew PD. Liver X receptor and retinoid X receptor agonists inhibit inflammatory responses of microglia and astrocytes. J. Neuroimmunol., 183, 50–59 (2007).
- 57) Ghisletti S, Huang W, Ogawa S, Pascual G, Lin ME, Willson TM, Rosenfeld MG, Glass CK. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARγ. Mol. Cell, 25, 57–70 (2007).
- 58) Orihuela R, McPherson CA, Harry GJ. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol., 173, 649–665 (2016).
- 59) Feng X, Ji Y, Zhang C, Jin T, Li J, Guo J. CCL6 promotes M2 polarization and inhibits macrophage autophagy by activating PI3-kinase/Akt signalling pathway during skin wound healing. Exp. Dermatol., 32, 403–412 (2023).
- 60) Franco R, Fernández-Suárez D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol., 131, 65–86 (2015).
- 61) Chen S, Saeed AFUH, Liu Q, Jiang Q, Xu H, Xiao GG, Rao L, Duo Y. Macrophages in immunoregulation and therapeutics. Signal Transduct. Target. Ther., 8, 207 (2023).
- 62) Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, Szeles L, Poliska S, Oros M, Evans RM, Barak Y, Schwabe J, Nagy L. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity, 33, 699–712 (2010).
- 63) Daniel B, Nagy G, Czimmerer Z, Horvath A, Hammers DW, Cuaranta-Monroy I, Poliska S, Tzerpos P, Kolostyak Z, Hays TT, Patsalos A, Houtman R, Sauer S, Francois-Deleuze J, Rastinejad F, Balint BL, Sweeney HL, Nagy L. The nuclear receptor PPARγ controls progressive macrophage polarization as a ligand-insensitive epigenomic ratchet of transcriptional memory. Immunity, 49, 615–626.e6 (2018).
- 64) Munder M. Arginase: an emerging key player in the mammalian immune system. Br. J. Pharmacol., 158, 638–651 (2009).