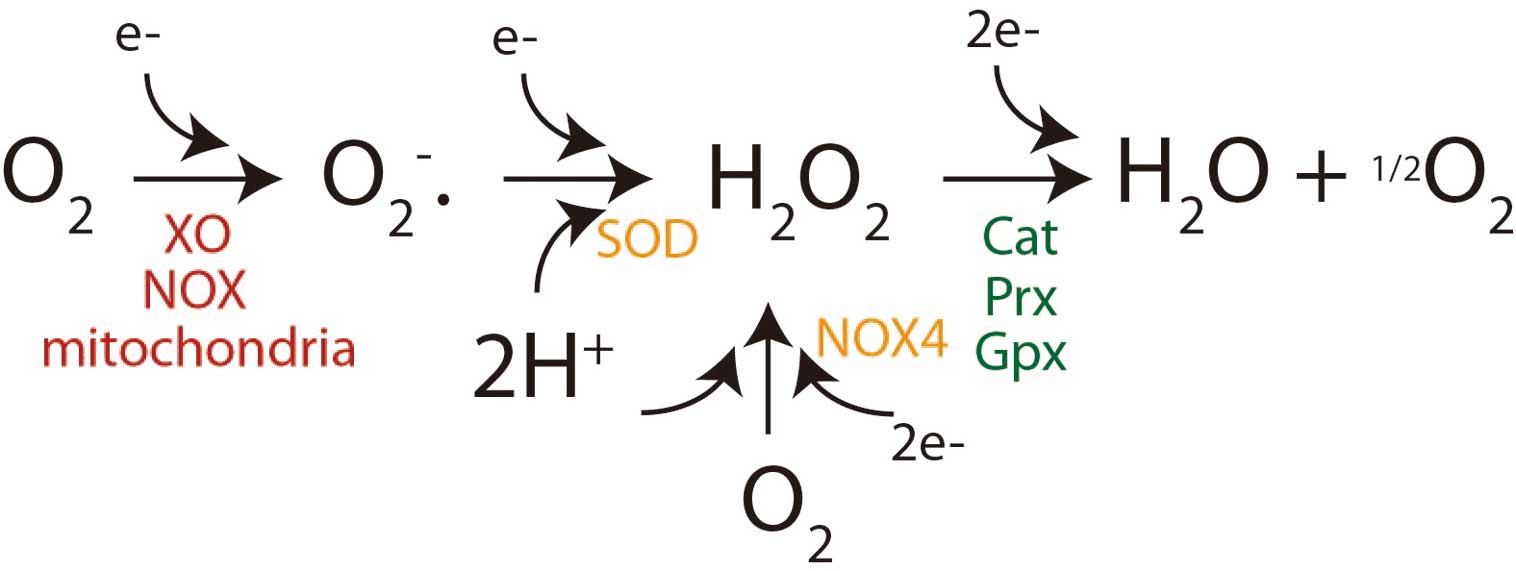
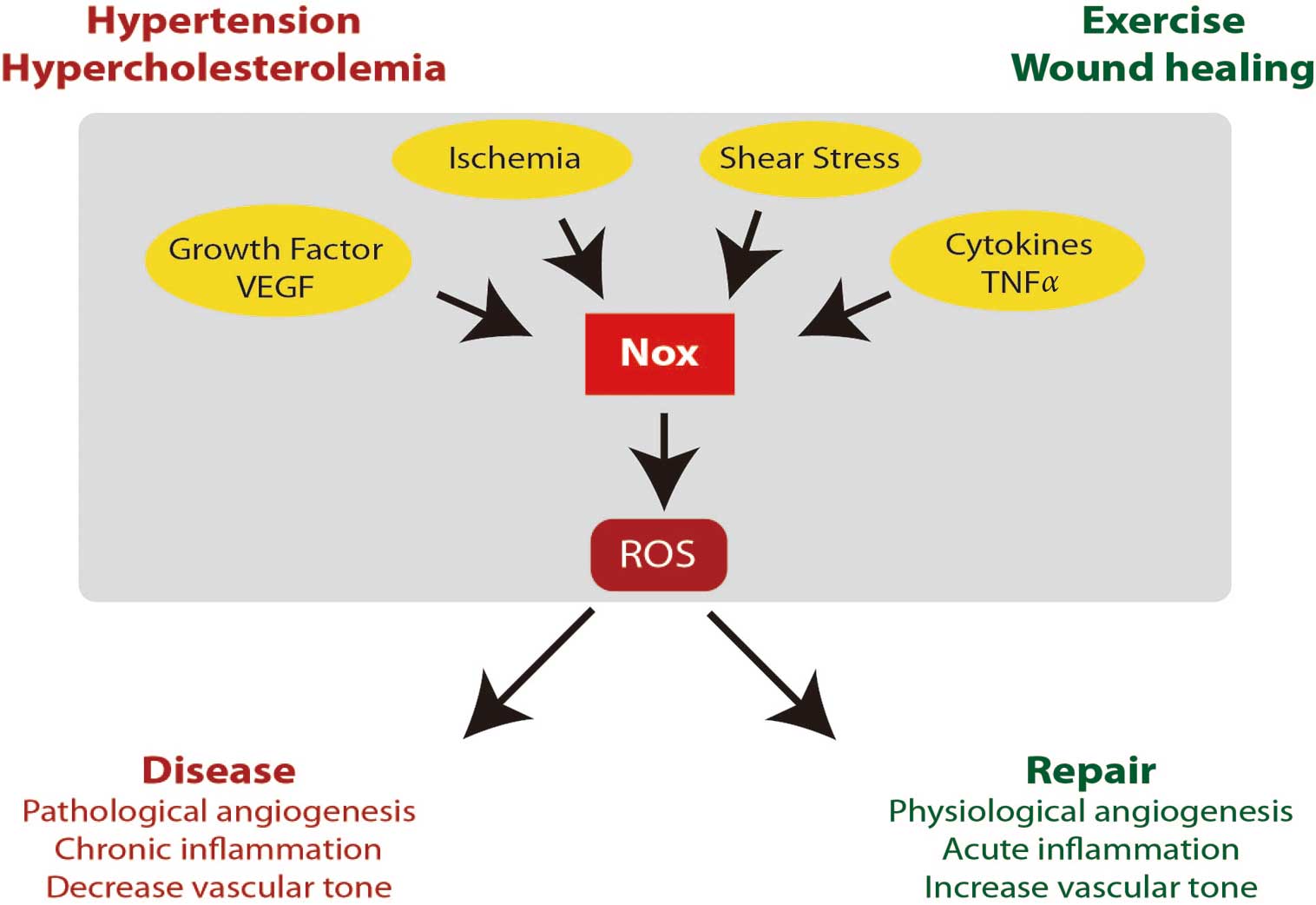
ROS in Endothelial Function and Dysfunction
Vascular Tone
The endothelium, in large part, controls vascular homeostasis, including the vascular tone of both conduit and resistance vessels. The ability of the endothelium to influence vascular tone is fundamental to the regulation of blood flow in response to many physiologic conditions. For example, muscular exercise produces an acute requirement for increased delivery of oxygen and nutrients that is largely achieved via dilation of resistance vessels to increase blood flow. Sustained increases in blood flow also increase the shear forces on the endothelium, thus prompting the release of vasodilators that result in conduit vessel dilation and normalization of endothelial shear. Conversely, tissue injury can prompt vasoconstriction as a means of stemming blood loss, a response that is, in part, due to the endothelium. These changes in vascular tone occur through the paracrine action of endothelial-derived vasorelaxants (eg, NO•, prostacyclin, and endothelial-derived hyperpolarizing factor [EDHF]) or vasoconstrictors (eg, leukotrienes and endothelin-1).29
In most cases, vascular production of ROS has proven important in the regulation of vascular tone (for reviews see Chatterjee et al,30
Brandes et al31) (Figure 2).
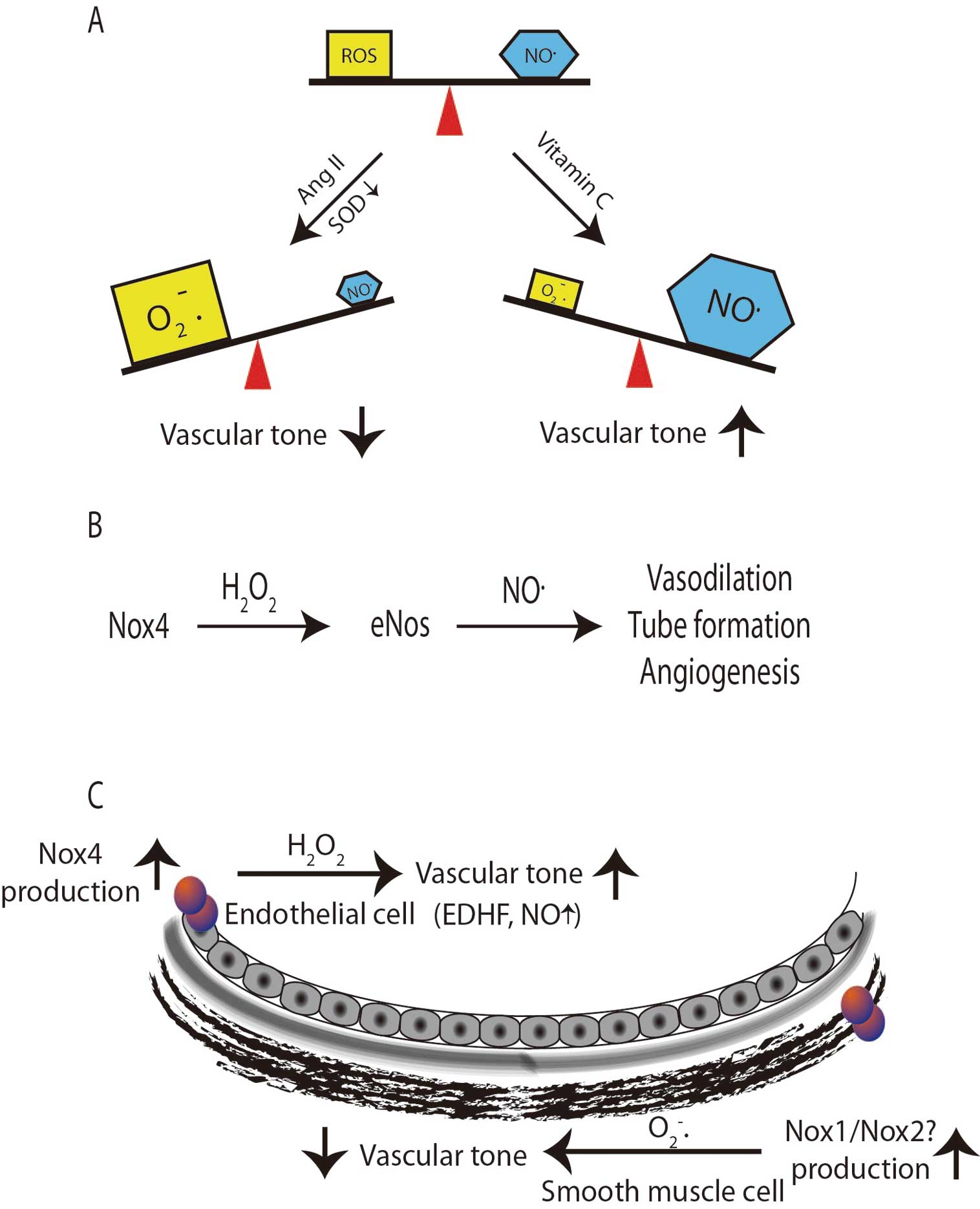
The effect of ROS on the control of vascular tone has been examined extensively in both human subjects and model animal systems. Human studies have largely focused on small molecules that affect ROS such as vitamin C. The studies used flow-mediated dilation as an index of endothelial NO•
regulation of vascular tone, and targeting ROS with vitamin C improved vascular function in a number of conditions known to be associated with excess oxidative stress (eg, type II diabetes and coronary artery disease).32–34
These data, and others, were interpreted to indicate that O2•−
production in disease states modifies vascular tone. More direct support for this idea came from animal studies in which SOD was inactivated and resulted in decreased vessel relaxation35
and endothelial dysfunction (Figure 2A).
Another consequence of vascular tone is the regulation of total peripheral resistance, which partially dictates blood pressure. Impaired control of resistance vessel function has been linked to hypertension and there is ample evidence that ROS play a role in this disorder. Understanding the potential mechanism(s) of hypertension is clearly important because it is present in up to 30% of the US population and affected individuals are at increased risk for stroke, aneurysm, coronary artery disease, peripheral artery disease and chronic kidney disease. Of interest, a role for ROS in hypertension was first introduced in the 1960 s36
and since this first demonstration, multiple cell, animal, and clinical studies have demonstrated that hypertension is associated with an increase in oxidative stress in part via NADPH oxidases. Studies in humans indicate that acute administration of vitamin C results in blood pressure reduction,37
suggesting an important role for ROS and endothelial dysfunction in this context. In model systems of hypertension, a strong link between blood pressure regulation and NADPH oxidase activity has been established. For example, angiotensin II (AngII) infusion produces hypertension and this process is linked to increased Nox-derived O2•−
production throughout the vascular wall in rats, leading to endothelial dysfunction.38
The infusion of AngII has been shown to increase the expression of multiple Nox proteins including Nox1, Nox2, Nox4, and p22phox.38
Over the ensuing decades, subsequent studies in humans and animal models of hypertension have been consistent in providing a link between Nox and hypertension.39
This link between NADPH oxidases and hypertension has been studied via genetic manipulation of Nox isoform expression and these studies have demonstrated that maladaptive effects of excess Nox activity are likely related to specific tissues and specific Nox family members. For example, Nox1 overexpression in vascular smooth muscle cells leads to an increase in hypertension and endothelial dysfunction in response to AngII.40,41
Conversely, deletion of Nox1 appears to prevent AngII-induced hypertension and also limits the development of endothelial dysfunction and oxidative stress.42–44
Infusion of AngII into Nox2-deficient animals had a minor effect on vascular hypertrophy, but very little effect on the development of hypertension.45,46
Examination of mice lacking Nox4 revealed them to be normotensive.
Most of the studies were focused on the Nox isoforms that generate O2•−, which is known to impair responses that are dependent on NO•
bioactivity. Perhaps focusing on specific reactive species could yield additional insight into how ROS affect vascular tone. In particular, H2O2
is distinct from O2•−
by not directly interacting with NO•. Moreover, it is known to stimulate guanylyl cyclase directly and induce vasodilation.47,48
These previous observations suggest that H2O2
can act as a vasodilator compound and have led many to conclude it is an EDHF as well.49
This has been substantiated in animal and in vitro models that involve the overexpression of Nox4 (considered a H2O2
generator) in the endothelium. In those studies, excess peroxide production was associated with an enhanced endothelial response to acetylcholine and a reduction in basal blood pressure that was normalized by supplementation with antioxidants.50
In light of these observations, coupled with the in vitro data demonstrating that H2O2
enhances eNOS expression and NO•
bioactivity,12,13
it is likely that H2O2
acts not only as an EDHF, but may also promote bioavailable NO•
in vivo. This notion is supported by studies in which Nox4 overexpression increased bioavailable NO•, whereas AngII infusion in animals lacking Nox4 resulted in decreased eNOS expression and NO•
production14,15
(Figure 2B). This further emphasizes that not only the location of ROS production, but also the precise species formed, is contextually important. Thus, we might consider that O2•−
produced within the smooth muscle cells leads to a hypertrophic, hypertensive phenotype, but H2O2
formed and excreted from endothelial cells enhances vascular tone (Figure 2C).
Inflammation/Immune Function
In addition to vascular tone, endothelial cells are key regulators of the local inflammatory and immune responses in both the vasculature and the surrounding tissues. Over the past 3 decades, atherosclerosis research has highlighted a deleterious role for ROS in the pathogenesis of the disease, which serves as a general model of inflammation. A paradigm generally agreed upon in the literature is depicted in
Figure 3A, which begins with an initial endothelial activation that can be precipitated by consuming an atherogenic diet for as little as a few days.51–54
In this model, endothelial activation is manifest, given the increased expression of leukocyte adhesion molecules and inflammatory cytokines that are responsible for the rolling, firm attachment, and diapedesis of inflammatory cells into the vascular wall. This is followed by the promotion of lipoprotein deposits in the subintimal space, retention of inflammatory cells (including monocytes), and ultimately foam cell formation. Continued cycles of lipid deposition, inflammatory cell infiltration and activation, and foam cell formation, lead to progressive formation of more complex atherosclerotic plaques that often involve a necrotic core covered by a thin fibrous cap separating the lesion from the circulation. Eventually, continued inflammation can weaken the fibrous cap, leading to its rupture and the formation of an occlusive thrombus that can precipitate acute events such as myocardial infarction, ventricular fibrillation, or stroke.
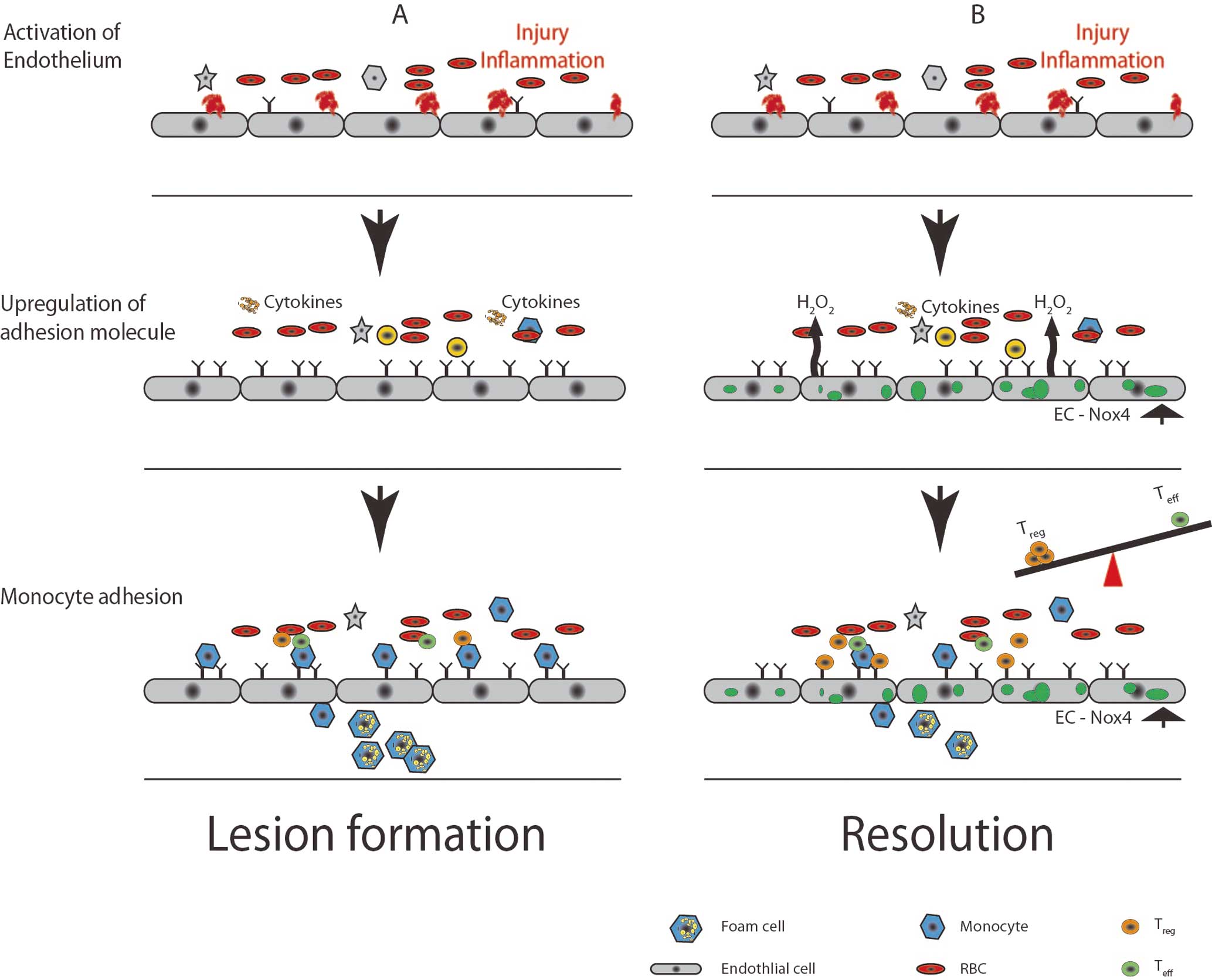
Multiple studies have documented increased Nox in the vasculature with atherosclerosis.28,55–59
It follows that atherosclerotic lesions in coronary arteries contain abundant p22phox and Nox2, which correlates with the severity of atherosclerosis.28,60
Similarly, vascular tissue from bypass patients with diabetes (a risk factor for atherosclerosis) demonstrated increased expression of p22phox, p47phox, and p67phox (all Nox binding partners) compared with nondiabetic subjects.61,62
In primates, the onset and progression of atherosclerosis is associated with increased Nox expression, whereas atherosclerosis regression is associated with a decrease in vascular Nox expression.59
Moreover, statins, which are known to decrease Nox expression, also decrease atherosclerosis,63–66
suggesting that Nox proteins may be involved in disease onset and progression.
ROS are important in endothelial activation, a phenomenon that is characterized by upregulation of adhesion molecules, increased vascular permeability, and reduced bioavailable NO•.67,68
Endothelial treatment with the cytokine tumor necrosis factor α (TNFα) produces increased adhesion molecules (ICAM1 and VCAM1) and chemokine (MCP1) expression via activation of Nox2.69–71
Endothelial lipopolysaccharide (LPS) exposure induces increases in adhesion molecules through Nox4 activation of TLR4, which leads to increased monocyte adhesion and transmigration.72
As mentioned before, AngII, a potent stimulator of Nox-derived ROS and AngII, also increases the expression of adhesion molecules on the endothelium through ROS production.73,74
Furthermore, aldosterone may involve Nox4 activation by the mineral corticoid receptor in its impairment of endothelial function.75
Oscillatory shear stress leads to endothelial activation, increased adhesion molecules, and matrix metalloproteinase (MMP) secretion through Nox.76
Bone morphogenic proteins (BMPs) also activates endothelial cells through ROS-dependent mechanisms,77
although the precise pathways remain unclear.
Although there is an abundance of data implicating Nox in the pathogenesis of atherosclerosis, data demonstrating a clear causal role for NADPH oxidases in atherosclerosis has proven difficult because studies in genetic mouse models have produced inconsistent results. For example, mice lacking Nox2 did not exhibit a significant difference in atherosclerotic lesion formation when looking exclusively at the aortic sinus.78
However, a more recent study examining the length of the aorta from the arch to the iliac bifurcation demonstrated that Nox2 expression precedes lesion development and that deletion of Nox2 protects from lesion deposits.56
In mice lacking the Nox accessory protein, p47phox, one study documented no difference in aortic root atherosclerosis,79
but another study demonstrated a significant protection in the descending aorta.80
The data for Nox1 are a little more consistent; Nox1-deficient mice mice crossed onto an ApoE−/−
model demonstrated decreased lesion formation57
and administration of the Nox1/4 inhibitor (GKT137831) in diabetic ApoE−/−
mice produced decreased lesion formation compared with controls, which was caused by Nox1 inhibition. Together these data indicate that more comprehensive studies are needed regarding the effect of Nox on location-specific promotion and progression of atherosclerosis.
More recent atherosclerosis research has focused on the later stages of the disease, which can involve not only the promotion of inflammation, but also its resolution. Among the regulators of inflammation that have garnered attention are regulatory T cells (Tregs). Tregs are an important subset of T cells that lend themselves to immune tolerance, and data indicate that Tregs are relevant for atherosclerosis. For example, Tregs are important in several processes: they inhibit pro-atherogenic T cells (Th1 and Th17; T effector) via cell-cell contact and cytokine secretion (IL10, transforming growth factor [TGF]β, IL35); they inhibit foam cell formation via downregulation of CD36; they suppress endothelial cell activation through cytokine secretion or reducing leukocyte adhesiveness; and they inhibit inflammatory macrophages by promoting polarization towards the M2 (ie, reparative) macrophage variety. Indeed, multiple studies have demonstrated an inverse correlation between FoxP3 (a main transcription factor in determining Treg fate) and atherosclerosis.81–86
There are recent data suggesting a link between ROS and Treg cells wherein commitment of the Treg lineage is dependent on localized production of ROS and that scavenging of ROS decreases the Tregs/Teffector balance. The majority of these studies have focused on intracellular T-cell ROS, but it has also been demonstrated that localized macrophage ROS production can determine T-cell fate, thus potentiating Treg cell production.87–90
As the endothelium is a major regulator of local inflammatory and immune responses, and predominantly secretes ROS into the extracellular space, one might speculate that endothelial-derived ROS could influence T-cell fate by potentiating Treg differentiation (Figure 3B). Definitive answers will require investigation with atherosclerosis models that involve manipulation of endothelial ROS sources.
Angiogenesis
Another major function of the endothelium is the initiation and promotion of angiogenesis, the process of new blood vessel formation. In the postnatal state, angiogenesis is largely in response to inadequate tissue blood flow resulting in localized tissue hypoxia. During ischemic conditions, decreased blood flow leads to decreased nutrient availability (eg, oxygen and glucose). These metabolic perturbations trigger the activation of HIF-1α and other transcription factors to coordinate the release of cytokines and growth factors such as vascular endothelial growth factor (VEGF), PDGF, angiopoietins, and TGFβ. These transcriptional changes are known to occur in multiple tissue types (eg, macrophages, fibroblasts, endothelial cells, keratinocytes).91–94
Both physiological angiogenesis (wound healing, vessel damage, ischemic repair) and pathological angiogenesis (tumors, diabetic retinopathy, age-related macular degeneration) involve the same initial signaling paradigms, all of which involve ROS.
Antioxidant-mediated ROS scavenging results in decreased angiogenesis in multiple models95–99
that could be considered in the setting of pathological angiogenesis. However, many studies have now demonstrated that physiological angiogenesis also requires ROS production. Utilizing a model of hindlimb ischemia for peripheral vascular repair, multiple groups have demonstrated that endothelial Nox-derived H2O2
production is an important component of angiogenesis and tissue repair.14,49,100,101
These data suggest that under certain circumstances, H2O2
is not a mediator of tissue injury, but rather an important component of the resolution of tissue damage.
Endothelial cells typically reside in tissues in a quiescent state and become activated in the setting of angiogenesis. One factor critically responsible for endothelial cell activation is VEGF and this process is, in part, ROS-dependent. For example, ROS upregulate VEGF expression via activation of HIF-1α.102–104
VEGF has multiple receptors, but binding to VEGF receptor 2 (VEGFR2) is of particular interest because it induces ROS production that appears critical for key angiogenesis processes such as basement membrane degradation, migration, proliferation, and tube formation (Figure 4).
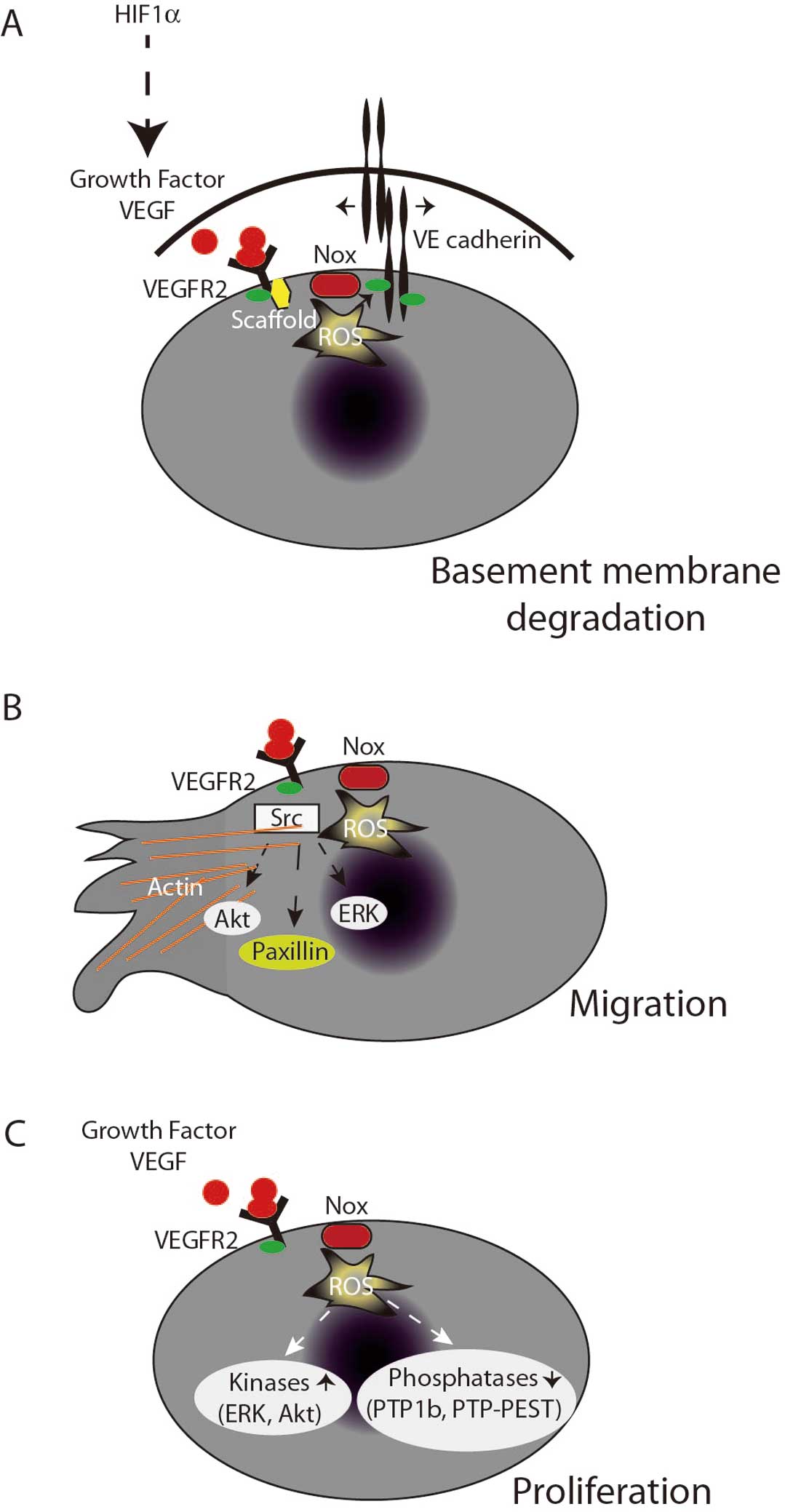
The process of endothelial cell migration and early tube formation requires initial detachment of the endothelial cell from its basement membrane followed by actin cytoskeletal rearrangements to facilitate movement, which also requires clearing out of extracellular matrix.105,106
These processes are tightly linked to signaling events in response to VEGF and angiopoietin-1.107–109
In quiescent endothelial cells, VE-cadherin dimers bind catenins anchoring the extracellular matrix to the actin cytoskeleton. Binding of VEGF to the VEGFR2 (flk-1/kdr) initiates binding of the scaffold IQGAP1 to actin, β-catenin, and Rac1, which then tightly associates with Ve-cadherin and the VEGFR.107,110,111
This process is critical for endothelial cell detachment and is characterized by localized ROS production that facilitates the tyrosine phosphorylation of VE-cadherin and β-catenin that is critical for disassembly of the VE-cadherin-catenin complex and junctional breakdown of the endothelial monolayer (Figure 4A).112
Endothelial cells must also reorganize the cytoskeleton to prepare for forward movement. Migrating endothelial cells create focal complexes transiently at their leading edge, which is critical for the cytoskeletal reorganization needed to produce filopodia or lamellipodia. In 2000, Moldovan et al describe discrete ROS formation at the leading edge of these membrane ruffles.113
Mechanistically, this occurs through IQGAP1 tethering Nox2 to actin at the leading edge110
where WAVE1 then recruits p47phox and binds Rac1, leading to membrane ruffle formation.114
This process also involves TRAF4, a protein known to directly bind p47phox,115
which directs oxidant production to the protein tyrosine phosphatase PTP-PEST and enhances focal complex phosphorylation,116
leading to directional migration. Thus, many of the steps involved in endothelial cell migration are ROS-dependent (Figure 4B).
During the process of angiogenesis, endothelial cells need to rapidly proliferate, and proliferating endothelial cells have increased ROS production as compared with quiescent cells.117
Moreover, Nox4-generated H2O2
is linked to the proliferative rate of endothelial cells.118
When exposed to growth factors (ie, serum), endothelial cell proliferation and activation downstream kinases (p38, ERK, and Akt) appear to be ROS-dependent and both Nox2 and Nox4 have been implicated in this process (Figure 4C).14,15,117,119,120
Finally, the migration and proliferation of endothelial cells ultimately results in tube formation, the earliest stage of new vessel formation. A recent observation indicates that autophagy, a process known to require ROS,121
is important in endothelial behavior relevant to tube formation. For example, ROS-driven autophagy results in endothelial survival in response to stressors such as caloric restriction.122
In addition, ROS-mediated autophagy is a key element in phenotypic responses of the endothelium critical for tube formation.123,124
Another important component of the tissue response to hypoxia is the dilation and recruitment of preformed capillaries to increase tissue blood flow. This process appears to be critical for the early responses to tissue ischemia and is enhanced by repeated metabolic stress such as exercise. Emerging data suggest that tissue ROS production is required for adaptation to exercise. For instance, exercise training increases blood flow, in part through increased expression of eNOS125
that can be recapitulated in vitro by H2O2. Exercise also increases tissue capillary density126–128
and tissue capillary density appears to be ROS-dependent,129,130
with at least one study specifically implicating Nox4.131