Abstract
Background:
In children with long QT syndrome (LQTS), risk factors for cardiac events have been reported, but age-, gender- and genotype-related differences in prognosis remain unknown in Asian countries.
Methods and Results:
The study examined clinical prognosis at age between 1 and 20 years in 496 LQTS patients who were genotyped as either of LQT1–3 (male, n=206). Heterozygous mutations were observed in 3 major responsible genes:
KCNQ1
in
271,
KCNH2
in 192, and
SCN5A
in 33 patients. LQTS-associated events were classified into 3 categories: (1) syncope (n=133); (2) repetitive torsade de pointes (TdP, n=3); and (3) cardiopulmonary arrest (CPA, n=4). The risk of cardiac events was significantly lower in LQT1 girls than boys≤12 years (HR, 0.55), whereas LQT2 female patients ≥13 years had the higher risk of cardiac events than male patients (HR, 4.60). Patients in the repetitive TdP or CPA group included 1 LQT1 female patient, 1 LQT2 male patient, and 5 LQT2 female patients. All LQT2 patients in these groups had TdP repeatedly immediately after the antecedent event. In addition, all 5 female LQT2 patients in these groups had the event after or near puberty.
Conclusions:
Female LQT2 children might have repeated TdP shortly after prior events, especially after puberty. (Circ J 2016; 80: 696–702)
Long QT syndrome (LQTS) is an inherited heart disease associated with increased propensity to syncope, torsade de pointes (TdP), and sudden arrhythmic death.1
It is an important cause of sudden cardiac death in children without structural heart disease.2
Syncope in LQTS is due to TdP, and death is usually due to ventricular fibrillation.3
Hundreds of mutations have been identified in >10 LQTS-susceptibility genes and approximately 75% of clinically diagnosed patients with LQTS are successfully genotyped. Most of them are found to carry a mutation in either one of 3 major genes:
KCNQ1
(LQT1),
KCNH2
(LQT2), or
SCN5A
(LQT3).4–6
Editorial p 598
Risk stratification depends on genotype, mutation type, and mutation location. Specifically, patients with cytoplasmic-loop (C-loop) missense
KCNQ1
and S5-loop-S6 missense
KCNH2
mutations have a greater risk for cardiac events.7–9
In neonates and infants, several previous studies have reported genetic background underlying fatal arrhythmias and indicated high prevalence of
SCN5A
and
KCNH2
(S5-loop-S6 missense) mutations.10–13
Risk factors for cardiac events in children from birth to 20 years have been reported: LQT1 boy ≤15 years, LQT2 girl ≥16 years, corrected QT interval (QTc) prolongation >500 ms, and history of prior syncope.14–17
Age-, gender- and genotype-related differences in prognosis, however, remain unknown in Asian countries, although the presence of country-specific hot spots in LQTS mutations has been reported.18
In addition, differences in clinical course shortly after the prior event remain unknown. Therefore, the present study focused on patients with symptomatic LQTS who were identified as mutation carriers in any of the 3 major genes responsible for LQTS in children (from 1 to 20 years), and analyzed their distribution and severity of disease.
Methods
Subjects
The study consisted of 496 LQTS patients (283 probands and 213 family members) who were genetically confirmed as either LQT1, LQT2, or LQT3. All but 1 Vietnamese boy were Japanese. They were referred to 2 institutes for genetic testing: Shiga University of Medical Science or Kyoto University Graduate School of Medicine between 1996 and December 2013. The study protocol was approved by the institutional review boards. Patients with compound or homozygous mutations were excluded from the study. We also excluded those with mutations in other LQTS-related genes. On enrollment, complete history was obtained retrospectively from birth to age of study entry. If someone was enrolled after reaching 20 years, we used only the clinical history from birth to 20 years. Patients who had received β-blockers before cardiac events were excluded during the analysis of the natural progress of the disease. No female patient was pregnant or in the postpartum phase when they had the first cardiac event. Thus, we assessed age- and genotype-related severity of LQTS children from 1 to 20 years.
LQTS-related events were classified into 3 categories: (1) syncope (transient and complete loss of consciousness); (2) repetitive TdP (documented within 24 h from the first syncope); and (3) cardiopulmonary arrest (CPA). The CPA group consisted of patients with aborted cardiac arrest requiring external defibrillation as part of resuscitation. In order to measure QTc (with Bazett correction19), 12-lead electrocardiograms (ECG) recorded at the enrolled age were used for asymptomatic patients. If asymptomatic patients were aged >20 years when enrolled, the first recorded ECG were used. For symptomatic patients, ECG recorded at onset were used. If there was no ECG available at onset, the ECG first recorded after onset was used. In patients with CPA, the ECG recorded before the cardiac event or ≥1 month after was used in order to avoid the influence of resuscitation.
Genetic Analysis
Screening for mutations was routinely performed for
KCNQ1,
KCNH2,
SCN5A,
KCNE1,
KCNE2, and
KCNJ2
using polymerase chain reaction (PCR) and denatured high-performance liquid chromatography (WAVE system; Transgenomic Omaha, NE, USA). For aberrant PCR products, subsequent direct DNA sequencing was conducted using a DNA sequencer (ABI 3130 DNA Sequencer, Perkin Elmer, Foster City, CA, USA). Genetic mutations were characterized by their location and coding effects on the amino acid sequence. We divided coding effects into 2 categories: (1) missense or (2) non-missense (putative splice sites, in-frame insertions/deletions, nonsense, and frameshift mutations). The C-loop region of
KCNQ1
was defined as the coding sequence involving amino acid residues 171–195 (S2–S3) and 242–262 (S4–S5), and the S5-loop-S6 region of
KCNH2
was defined as the amino acid region ranging from residue 552 to 657.
Statistical Analysis
Statistical analysis was performed using SPSS 22.0 (IBM, Armonk, NY, USA). Data are given as mean±SD for continuous variables and number (percentage) for categorical variables. Differences in characteristics were evaluated using unpaired Student’s t-test, Mann-Whitney U-test, or chi-squared test, as appropriate. Then, we analyzed patients with
KCNQ1
and
KCNH2
because of the limited number of the patients with
SCN5A. We used the Kaplan-Meier method to estimate the distribution of time to first cardiac event before β-blocker therapy based on genotype-gender subsets (categorized as male with
KCNQ1
mutation, female with
KCNQ1
mutation, male with
KCNH2
mutation, and female with
KCNH2
mutation), using the log-rank test to compare differences between subsets. Cox proportional hazards models, allowing for separate baseline hazard function for each genotype, were used to assess the association of gender (female vs. male) with first cardiac event within each genotype and age group, adjusted for QTc.
The risk of cardiac events may be related to sex hormone factors.1
Puberty begins with the elevation of sex hormones, especially estrogen in girls. The mean age of puberty onset is 12.5±0.9 years in boys and 10.0±1.4 years in girls.20
In addition, menarche begins at the mean age of 12.4±1.0 years,21
after that time girls have higher estrogen to progesterone ratio around ovulation. Therefore, we conducted separate Cox models for 2 age groups: age 1–12 years and age 13–20 years. One year of age was used as the time origin in the analysis of mutation carriers age 1–12 years, whereas for the mutation carriers aged 13–20 years, 13 years of age was considered as the time origin. Differences were accepted as statistically significant for 2-tailed P<0.05.
Results
Clinical Characteristics
Table 1
lists clinical characteristics according to genotype and gender. LQT1 or LQT2 patients had no gender-related difference in QTc in the 1–12-year age group (LQT1, P=0.73; LQT2, P=0.11). In contrast, in the ≥13-year age group, girls had significantly longer QTc than boys both in the LQT1 (male, 445±38 ms; female, 471±43 ms; P<0.001) and the LQT2 groups (male, 478±63 ms; female, 505±51 ms; P=0.002). Compared for age in gender-genotype, QTc was shorter in LQT1 male patients ≥13 years than in those ≤12 years (445±38 ms vs. 475±36 ms, P<0.001), and the QTc was longer in LQT2 girls ≥13 years than in those ≤12 years (505±51 ms vs. 474±35 ms, P<0.001), although there were no age-dependent differences in LQT1 girls (P=0.47) or LQT2 boys (P=0.23). LQT3 patients had no gender-related or age-dependent difference in QTc.
Table 1.
Subject Clinical Characteristics vs. Genotype and Gender
| |
KCNQ1 mutation carrier (n=271) |
KCNH2 mutation carrier (n=192) |
SCN5A mutation carrier (n=33) |
Male
(n=104) |
Female
(n=167) |
Male
(n=81) |
Female
(n=111) |
Male
(n=21) |
Female
(n=12) |
| Mean follow-up (years) |
10
(7–15) |
14
(9–20)** |
15
(10–20) |
16
(12–20) |
14
(12–20) |
13
(11–20) |
| ECG data (without β-blocker) |
| ECG recorded at ≤12 years |
n=52 |
n=47 |
n=24 |
n=24 |
n=8 |
n=4 |
| Age at ECG (years) |
8
(6.2–10.7) |
7
(7–10) |
9
(7.2–11) |
7.5
(6–9.7) |
11
(7.5–12) |
8.5
(6–11.7) |
| RR (ms) |
840
(730–979) |
821
(769–893) |
833
(713–985) |
783
(687–910) |
799
(777–951) |
751
(543–1,160) |
| QTc (ms) |
470
(452–493)††† |
475
(460–488) |
496
(454–533) |
472
(448–494) |
463
(416–499) |
438
(389–495) |
| QTc >500 ms |
12 (23) |
8 (17) |
11 (45) |
5 (20) |
2 (25) |
1 (25) |
| ECG recorded at ≥13 years |
n=46 |
n=103 |
n=50 |
n=77 |
n=12 |
n=7 |
| Age at ECG (years) |
30.5
(15–45) |
36
(17–42) |
29
(16–52.2) |
30
(17–40.5) |
32
(14–43) |
47
(16–58) |
| RR (ms) |
959
(880–1,057)†† |
967
(866–1,090)†† |
1,005
(905–1,152)*,†† |
921
(823–1,044)†† |
1,000
(843–1,091)† |
1,100
(846–1,170) |
| QTc (ms) |
448
(419–464) |
463
(444–484)*** |
476
(443–500) |
503
(469–526)**,††† |
422
(377–484) |
483
(427–495) |
| QTc >500 ms |
4 (8) |
18 (17) |
16 (28) |
45 (51)** |
2 (15) |
2 (25) |
| Cardiac events |
| Total |
30 (28) |
45 (26) |
16 (19) |
43 (38)** |
2 (9.5) |
4 (33) |
| Syncope |
30 (28) |
44 (26) |
15 (18) |
38 (34)* |
2 (9.5) |
4 (33) |
| Repetitive TdP |
0 |
0 |
0 |
3 (2) |
0 |
0 |
| CPA |
0 |
1 (0.5) |
1 (1) |
2 (1) |
0 |
0 |
Data given as median (IQR) or n (%). *P<0.05, **P<0.01, ***P<0.001 for comparison of male and female in the same genotype and age group. †P<0.05, ††P<0.01, †††P<0.001 for comparison of the groups ≤12 and ≥13 years in the same genotype and gender group. CPA, cardiopulmonary arrest; ECG, electrocardiogram; QTc, corrected QT interval; TdP, torsade de pointes.
In LQT2 patients, female subjects were more likely to have cardiac events between 1 and 20 years than male subjects (male, 16/81, 19%; female, 43/111, 38%; P=0.005), although there was no significant gender-related difference in the events rate in the LQT1 and LQT3 patients (LQT1, P=0.74; LQT3, P=0.16).
Age at first symptom onset and the respective number of genotyped patients are summarized in
Table 2. A total of 140 patients (28%) had cardiac events between 1 and 20 years of age, and the mutation distribution was as follows:
KCNQ1
mutations, n=75;
KCNH2
mutations, n=59; and
SCN5A
mutations, n=6. LQT2 patients were more likely to have CPA or repetitive TdP than LQT1 patients (LQT1, 1/271, 0.3%; LQT2, 6/192, 3%; P=0.022). There was no significant difference in QTc between the CPA (515±55 ms), repetitive TdP (554±84 ms), and syncope (491±54 ms) groups (P=0.22), and the QTc in syncope group patients was longer than in asymptomatic patients (470±45 ms, P<0.001;
Table 3).
Table 2.
Age at Symptom Onset vs. Genotype
| |
Total |
Male |
Female |
| n (%) |
Onset age
(years),
median
(IQR) |
n (%) |
Onset age
(years),
median
(IQR) |
n (%) |
Onset age
(years),
median
(IQR) |
Symptomatic/
Total |
CPA or
repetitive
TdP |
Symptomatic/
Total |
CPA or
repetitive
TdP |
Symptomatic/
Total |
CPA or
repetitive
TdP |
| KCNQ1 |
75/271 (27) |
1 (0.3) |
10 (7–11) |
30/104 (28) |
0 (0) |
7 (6–10) |
45/167 (26) |
1 (0.5) |
10 (8–13)** |
| Missense |
| C-loop |
11/41 (26) |
|
9 (7–13) |
6/15 (40) |
|
7 (5.7–9.7) |
5/26 (19) |
|
11 (9.5–14) |
| Non-C-loop |
44/169 (26) |
|
9 (7–11) |
16/62 (25) |
|
7 (6–10) |
28/107 (26) |
|
10 (7.2–11.7) |
| Non-missense |
20/61 (32) |
|
10 (7–14.5) |
8/27 (29) |
|
10 (5.2–10.7) |
12/34 (35) |
|
13 (7.7–16) |
| KCNH2 |
59/192 (30) |
6 (3)† |
13 (9–15)††† |
16/81 (19) |
1 (1.2) |
10 (7.2–12.7)† |
43/111 (38)** |
5 (4.5)† |
14 (12–16)**,†† |
| Missense |
| S5-loop-S6 |
19 /46 (41) |
|
12 (8–15) |
6/21 (28) |
|
10.5 (6.7–15.5) |
13/25 (52) |
|
13 (9–15) |
| Non-S5-loop-S6 |
17/67 (25) |
|
13 (11–16) |
4/26 (15) |
|
10.5 (6.2–12.5) |
13 /41 (31) |
|
14 (12–17) |
| Non-missense |
23/79 (29) |
|
14 (8.5–16) |
6/34 (17) |
|
9 (6.5–11.5) |
17/45 (37) |
|
15 (13–18)** |
| SCN5A |
6/33 (18) |
0 (0) |
14 (9.7–16.2) |
2/21 (9) |
0 (0) |
16, 17 |
4/12 (33) |
0 (0) |
11.5 (7.2–15) |
| Missense |
5/25 (20) |
|
12 (8.5–16.5) |
1/15 (6) |
|
17 |
4/10 (40) |
|
11.5 (7.2–15) |
| Non-missense |
1/8 (12) |
|
16 |
1/6 (16) |
|
16 |
0/2 (0) |
|
– |
| Total |
140/496 (28) |
7 (1.4) |
10.5 (7–14) |
48/206 (23) |
1 (0.4) |
9 (6–11) |
92/290 (31)* |
6 (2) |
12 (9–15)** |
*P<0.05, **P<0.01, ***P<0.001 for the comparison of male and female in the same genotype. †P<0.05, ††P<0.01, †††P<0.001 for the comparison of KCNQ1 and KCNH2 mutation carrier. C-loop, cytoplasmic loop. Other abbreviations as in Table 1.
Table 3.
Subject Characteristics vs. Clinical Symptom
| |
CPA
(n=4) |
TdP
(n=3) |
Syncope
(n=102) |
Asymptomatic
(n=345) |
| ECG data (without β-blocker) |
| RR (ms) |
690, 869, 967, 1,304 |
882, 1,132, 1,176 |
920 (800–1,037) |
920 (800–1,024) |
| QTc (ms) |
464, 472, 557, 566 |
473, 549, 640* |
480 (461–518)*** |
466 (446–493) |
| QTc >500 ms |
2 (50) |
2 (66) |
39 (38)* |
74 (21) |
| Onset age (years) |
8, 12, 13, 15 |
9, 14, 15 |
11.0±4.6 |
– |
Data given as n (%), mean±SD or median (IQR). *P<0.05, **P<0.01, ***P<0.001 compared with the asymptomatic group. Abbreviations as in Table 1.
All mutations were heterozygous and were classified into missense or non-missense. With regard to
KCNQ1
mutations, there were 11 missense in the C-loop, 57 missense in the non-C-loop, and 7 non-missense. Among
KCNH2
mutations, there were 19 missense in the S5-loop-S6, 17 missense in the non-S5-loop-S6, and 23 non-missense. There were 5 missense and 1 non-missense mutations in SCN5A (Table 2).
Genotype and Age at First Cardiac Event
As shown in
Table 2, mean onset age in the
KCNH2
group was significantly higher (12.5±4.7 years) than in the
KCNQ1
group (9.7±3.8 years, P<0.001), and this propensity was similar in both boys (KCNQ1, 8.1±3.3 years;
KCNH2, 9.8±3.9 years; P=0.028) and girls (KCNQ1, 10.7±3.8 years;
KCNH2, 13.2±4.7 years; P=0.002). Boys became symptomatic at a significantly younger age than girls in the
KCNQ1
(P=0.003) and
KCNH2
(P=0.005) groups. There were no significant differences in mean patient age according to type and location of mutation in
KCNQ1
and
KCNH2, respectively. Furthermore, the same pattern was observed in both boys and girls.
Age at onset was not significantly different between the CPA (12.0±3.0 years), repetitive TdP (12.7±3.3 years), and syncope (11.0±4.6 years) groups (P=0.73;
Table 3).
When comparing by genotype, the cumulative probability of a first cardiac event by age 20 years was 27% in LQT1, 30% in LQT2 and 18% in LQT3 (Figure 1).
Given that the number of
KCNQ1
and of
KCNH2
mutation carriers was relatively large, we examined the gender difference in these 2 groups (Figure 2).
Figure 2A
shows cumulative probability of a first cardiac event in patients with
KCNQ1
mutation. On Cox regression analysis, LQT1 girls ≤12 years had significantly lower risk for cardiac events than boys (HR, 0.55; 95% CI: 0.33–0.93; P=0.024), whereas there was no significant gender difference in risk in LQT1 patients ≥13 years (HR, 1.65; 95% CI: 0.36–7.54; P=0.53;
Table 4).
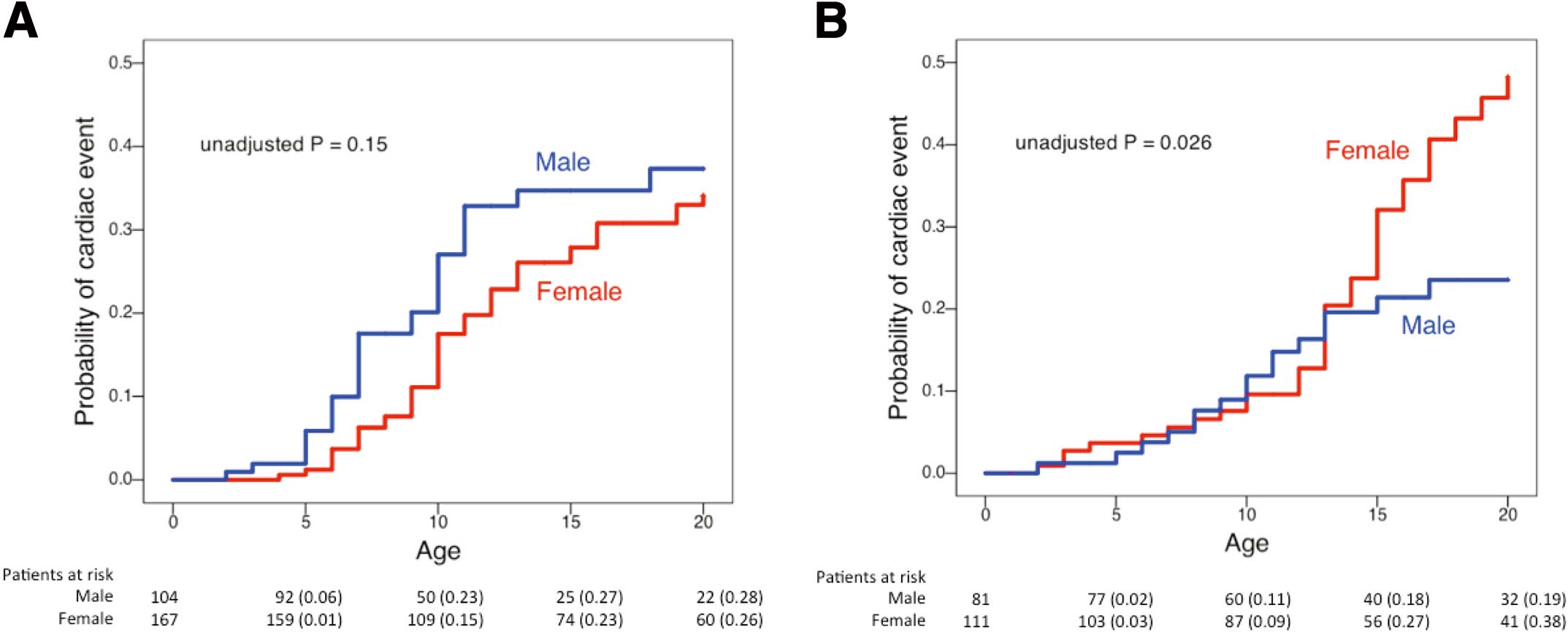
Table 4.
Significant Indicators of First Cardiac Event
| |
LQTS mutation carriers age 1–12 years |
LQTS mutation carriers age 13–20 years
without prior events |
| HR |
95% CI |
P-value |
HR |
95% CI |
P-value |
| Female vs. Male |
| KCNQ1 |
0.55 |
0.33–0.93 |
0.024 |
1.65 |
0.36–7.54 |
0.53 |
| KCNH2 |
0.71 |
0.32–1.55 |
0.39 |
4.60 |
1.62–13.07 |
0.004 |
| QTc (10-ms increase) |
1.002 |
0.998–1.007 |
0.24 |
1.012 |
1.008–1.016 |
<0.001 |
The Cox analysis involved 86 first cardiac events from 1 to 12 years in 463 KCNQ1 or KCNH2 mutation carriers, and 48 first cardiac events from 13 to 20 years in 263 KCNQ1 or KCNH2 mutation carriers without cardiac events before age 13. LQTS, long QT syndrome; QTc, corrected QT interval.
Figure 2B
shows cumulative probability of a first cardiac event in LQT2 patients. The Kaplan-Meier curve clearly shows an exponential increase in symptomatic female patients after puberty, resulting in a cross-over of the 2 curves. Furthermore, LQT2 girls ≥13 years had a significant increase in the risk compared with boys of the same age range (HR, 4.60; 95% CI: 1.62–13.07; P=0.004;
Table 4), although there was no significant gender difference in those ≤12 years (HR, 0.71; 95% CI: 0.32–1.55; P=0.39).
CPA and Repetitive TdP
Table 5
summarizes the genetic background in 7 children with CPA or repetitive TdP. Interestingly, their mutations were either in the channel-pore region or frameshift/nonsense in the C-terminus. There were 6 patients with LQT2 (5 girls) and only 1 with LQT1 (12-year-old girl). All these 6 female patients with severe cardiac events had onset after or near puberty. The only male patient was an 8-year-old boy with CPA (CPA case 2) who had hypokalemia (<3.0 mEq/L) due to severe diarrhea at the cardiac event.
Table 5.
CPA and Repetitive TdP: Genetic Background
| ID no. |
Gender |
Age at event
(years) |
Gene/Mutation |
RR (ms) |
QTc (ms) |
TdP† |
| CPA |
| 1 |
F |
12 |
KCNQ1 S277L |
967 |
464 |
(−) |
| 2 |
M |
8 |
KCNH2 K897fs+49X |
690 |
566 |
(+) |
| 3 |
F |
13 |
KCNH2 A561V |
869 |
472 |
(+) |
| 4 |
F |
15 |
KCNH2 K638del |
1,304 |
557 |
(+) |
| Repetitive TdP |
| 1 |
F |
9 |
KCNH2 S871fs+31X |
1,176 |
473 |
(+) |
| 2 |
F |
14 |
KCNH2 F640del |
882 |
640 |
(+) |
| 3 |
F |
15 |
KCNH2 L908fs+30X |
1,132 |
549 |
(+) |
†Documented within 24 h from the first cardiac event. F, female; M, male. Other abbreviations as in Table 1.
Discussion
In the present study, we found that the phenotype in LQT2 was more severe than in LQT1 (Table 2), although the overall event rates were similar (Figure 1). We also showed that female LQT2 patients had TdP repeatedly within a short term after prior events, especially after puberty. In a study on 1,520 patients with LQTS before the age of 40 years, documented TdP was reportedly more common among female than male patients, particularly among LQT2 mutation carriers.22
The previous study, however, did not mention whether the TdP occurred repeatedly immediately after the event or not in LQT2 patients. The present study first reported that repetitive TdP or fatal events were more common in LQT2. If a patient were suspected of LQT2 based on ECG findings (QT prolongation, notched, or bifid T waves), he/she would have repetitive events after the first TdP attack. In such a condition, genetic testing would be of clinical importance because the genotype (eg, mutations in the channel-pore region) appears to influence disease severity.
Female LQT2 patients have their first cardiac event at a higher rate after the onset of adolescence.17,23
Consistent with previous data, the present study noted a significantly prolonged QTc and increased risk of first cardiac event after puberty in LQT2 female patients, although there was no significant age-related difference in LQT2 male patients. Therefore, sex hormones may contribute to QT interval shortening or prolongation and the occurrence of cardiac events. In addition, hormonal state varies with the menstrual cycle in female patients after puberty.
In the menstrual cycle, longer QT interval is observed during the follicular phase than during the luteal phase.24
Although electrophysiological experiments using a guinea pig model showed that estrogen prolongs the action potential duration (APD) through the inhibition of
IKr,25
the effect of estrogen on QT interval in humans remains controversial.26
Progesterone shortens APD, which is mainly attributable to the enhancement of
IKs
under basal conditions and the inhibition of
ICaL
under cAMP stimulation, and a computer simulation model using a 50%
IKr
block indicated a protective effect of progesterone against arrhythmia.27
Furthermore, another study in an LQT2 rabbit model showed the proarrhythmic effect of estradiol and the anti-arrhythmic effect of progesterone.28
With regard to the 6 LQT2 patients in the CPA or repetitive TdP groups, all had TdP repeatedly shortly after the first cardiac event; moreover 4 of 6 were post-puberty girls. In the menstrual cycle, there are a number of days in which the estrogen:progesterone ratio is high, around ovulation.26
Although we were not able to check their menstrual phase, we suspected that LQT2 girls might have TdP repeatedly in the high estrogen:progesterone ratio phase. In contrast, an 8-year-old boy and a 9-year-old girl (Table 2) also had short-term TdP recurrence. Although the estrogen level might have begun to elevate in the 9-year-old girl, there may be other factors affecting the vulnerability to arrhythmias in LQT2 children.
In the CPA and repetitive TdP group, 1 patient had a missense mutation in the S5-loop-S6 region, 2 had a deletion mutation in the S5-loop-S6 region, and 3 had a frameshift/nonsense mutation in the C-terminus region. In line with the present findings, LQT2 patients with S5-loop-S6 missense mutations have been shown to have a high risk for cardiac events.8
In the
KCNH2
C-terminus regions, nonsense mutations were also reportedly more malignant than missense mutations.8
In the present LQT1 male cohort, the Kaplan-Meier curve shows that cardiac events obviously decreased after puberty (Figure 2A). Testosterone has been shown to shorten APD mainly through the enhancement of
IKs, in part due to
ICaL
suppression at higher concentrations.29
An adult cross-sectional study showed that the QTc interval was significantly shorter at high testosterone levels.30
Consistent with the previous report,17
increased testosterone may contribute to reduce the risk of cardiac event in male patients with
KCNQ1
mutation.
One of the limitations of the present study was that, because we analyzed 3 major LQTS genotypes, the contribution of other minor LQTS-related mutations to cardiac events could not be entirely excluded. In addition, patient data were collected at only 2 institutes, therefore there might be a bias in patient entry and genetic background, and hence a larger collaborative study is needed.
Conclusions
To the best of our knowledge, we report for the first time that female LQT2 patients are likely to have TdP recurrence shortly after antecedent cardiac events, especially after puberty. Therefore, short-term recurrence of TdP is expected and these patients should be followed more closely. These findings will help in selecting an appropriate preventive strategy based on the age of onset and genotype.
Acknowledgments
The study was supported in part by MEXT KAKENHI Grant Number 24591575 (to S.O.), 25136705, 25460406 (to H.I.), and 25461054 (to T.M.) from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, grants from the Ministry of Health, Labor and Welfare of Japan for Clinical Research on intractable Disease (H26-040, H24-033 to M.H.) and a Translational Research grant from the Japanese Circulation Society (to M.H.)
Disclosures
The authors declare no conflicts of interest.
References
- 1.
Goldenberg I, Moss AJ. Long QT syndrome. J Am Coll Cardiol 2008; 51: 2291–2300.
- 2.
Vincent GM. The Long QT and Brugada syndromes: Causes of unexpected syncope and sudden cardiac death in children and young adults. Semin Pediatr Neurol 2005; 12: 15–24.
- 3.
Roden DM. Clinical practice: Long-QT syndrome. N Engl J Med 2008; 358: 169–176.
- 4.
Tester DJ, Ackerman MJ. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation 2011; 123: 1021–1037.
- 5.
Shimizu W. Update of diagnosis and management of inherited cardiac arrhythmias. Circ J 2013; 77: 2867–2872.
- 6.
Mizusawa Y, Horie M, Wilde AA. Genetic and clinical advances in congenital long QT syndrome. Circ J 2014; 78: 2827–2833.
- 7.
Barsheshet A, Goldenberg I, O-Uchi J, Moss AJ, Jons C, Shimizu W, et al. Mutations in cytoplasmic loops of the KCNQ1 channel and the risk of life-threatening events: Implications for mutation-specific response to beta-blocker therapy in type 1 long-QT syndrome. Circulation 2012; 125: 1988–1996.
- 8.
Migdalovich D, Moss AJ, Lopes CM, Costa J, Ouellet G, Barsheshet A, et al. Mutation and gender-specific risk in type 2 long QT syndrome: Implications for risk stratification for life-threatening cardiac events in patients with long QT syndrome. Heart Rhythm 2011; 8: 1537–1543.
- 9.
Shimizu W, Moss AJ, Wilde AA, Towbin JA, Ackerman MJ, January CT, et al. Genotype-phenotype aspects of type 2 long QT syndrome. J Am Coll Cardiol 2009; 54: 2052–2062.
- 10.
Kato K, Makiyama T, Wu J, Ding WG, Kimura H, Naiki N, et al. Cardiac channelopathies associated with infantile fatal ventricular arrhythmias: From the cradle to the bench. J Cardiovasc Electrophysiol 2014; 25: 66–73.
- 11.
Cuneo BF, Etheridge SP, Horigome H, Sallee D, Moon-Grady A, Weng HY, et al. Arrhythmia phenotype during fetal life suggests long-QT syndrome genotype: Risk stratification of perinatal long-QT syndrome. Circ Arrhythm Electrophysiol 2013; 6: 946–951.
- 12.
Horigome H, Nagashima M, Sumitomo N, Yoshinaga M, Ushinohama H, Iwamoto M, et al. Clinical characteristics and genetic background of congenital long-QT syndrome diagnosed in fetal, neonatal, and infantile life: A nationwide questionnaire survey in Japan. Circ Arrhythm Electrophysiol 2010; 3: 10–17.
- 13.
Lupoglazoff JM, Denjoy I, Villain E, Fressart V, Simon F, Bozio A, et al. Long QT syndrome in neonates: Conduction disorders associated with HERG mutations and sinus bradycardia with KCNQ1 mutations. J Am Coll Cardiol 2004; 43: 826–830.
- 14.
Goldenberg I, Moss AJ, Peterson DR, McNitt S, Zareba W, Andrews ML, et al. Risk factors for aborted cardiac arrest and sudden cardiac death in children with the congenital long-QT syndrome. Circulation 2008; 117: 2184–2191.
- 15.
Liu JF, Jons C, Moss AJ, McNitt S, Peterson DR, Qi M, et al. Risk factors for recurrent syncope and subsequent fatal or near-fatal events in children and adolescents with long QT syndrome. J Am Coll Cardiol 2011; 57: 941–950.
- 16.
Hobbs JB, Peterson DR, Moss AJ, McNitt S, Zareba W, Goldenberg I, et al. Risk of aborted cardiac arrest or sudden cardiac death during adolescence in the long-QT syndrome. JAMA 2006; 296: 1249–1254.
- 17.
Zareba W, Moss AJ, Locati EH, Lehmann MH, Peterson DR, Hall WJ, et al. Modulating effects of age and gender on the clinical course of long QT syndrome by genotype. J Am Coll Cardiol 2003; 42: 103–109.
- 18.
Itoh H, Dochi K, Shimizu W, Denjoy I, Ohno S, Aiba T, et al. A common mutation of long QT syndrome type 1 in Japan. Circ J 2015; 79: 2026–2030.
- 19.
Bazett H. An analysis of the time relations of electrocardiograms. Heart 1920; 7: 353–370.
- 20.
Matsuo N. Skeletal and sexual mutation in Japanese children. Clin Pediatr Endocrinol 1993; 2: 1–4.
- 21.
Ashizawa K. Skeletal and sexual maturation and growth in Tokyo girls: Longitudinal observations. Clin Pediatr Endocrinol 1993; 2: 5–8.
- 22.
Locati EH, Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Lehmann MH, et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: Findings from the International LQTS Registry. Circulation 1998; 97: 2237–2244.
- 23.
Zareba W, Moss AJ, Schwartz PJ, Vincent GM, Robinson JL, Priori SG, et al. Influence of genotype on the clinical course of the long-QT syndrome: International Long-QT Syndrome Registry Research Group. N Engl J Med 1998; 339: 960–965.
- 24.
Nakagawa M, Ooie T, Takahashi N, Taniguchi Y, Anan F, Yonemochi H, et al. Influence of menstrual cycle on QT interval dynamics. Pacing Clin Electrophysiol 2006; 29: 607–613.
- 25.
Kurokawa J, Tamagawa M, Harada N, Honda S, Bai CX, Nakaya H, et al. Acute effects of oestrogen on the guinea pig and human IKr channels and drug-induced prolongation of cardiac repolarization. J Physiol 2008; 586: 2961–2973.
- 26.
Sedlak T, Shufelt C, Iribarren C, Merz CN. Sex hormones and the QT interval: A review. J Womens Health 2012; 21: 933–941.
- 27.
Nakamura H, Kurokawa J, Bai CX, Asada K, Xu J, Oren RV, et al. Progesterone regulates cardiac repolarization through a nongenomic pathway: An in vitro patch-clamp and computational modeling study. Circulation 2007; 116: 2913–2922.
- 28.
Odening KE, Choi BR, Liu GX, Hartmann K, Ziv O, Chaves L, et al. Estradiol promotes sudden cardiac death in transgenic long QT type 2 rabbit while progesterone is protective. Heart Rhythm 2012; 9: 823–832.
- 29.
Bai CX, Kurokawa J, Tamagawa M, Nakaya H, Furukawa T. Nontranscriptional regulation of cardiac repolarization currents by testosterone. Circulation 2005; 112: 1701–1710.
- 30.
Zhang Y, Ouyang P, Post WS, Dalal D, Vaidya D, Blasco-Colmenares E, et al. Sex-steroid hormones and electrocardiographic QT-interval duration: Findings from the third National Health and Nutrition Examination Survey and the Multi-Ethnic Study of Atherosclerosis. Am J Epidemiol 2011; 174: 403–411.


