Abstract
Antioxidants are expected to improve cardiovascular disease (CVD) by eliminating oxidative stress, but clinical trials have not shown promising results in chronic CVD. Animal studies have revealed that reactive oxygen species (ROS) exacerbate acute CVDs in which high levels of ROS are observed. However, ROS are also necessary for angiogenesis after ischemia, because ROS not only damage cells but also stimulate the cell signaling required for angiogenesis. ROS affect signaling by protein modifications, especially of cysteine amino acid thiols. Although there are several cysteine modifications, S-glutathionylation (GSH adducts; -SSG), a reversible cysteine modification by glutathione (GSH), plays an important role in angiogenic signal transduction by ROS. Glutaredoxin-1 (Glrx) is an enzyme that specifically removes GSH adducts in vivo. Overexpression of Glrx inhibits, whereas deletion of Glrx improves revascularization after mouse hindlimb ischemia. These studies indicate that increased levels of GSH adducts in ischemic muscle are beneficial in promoting angiogenesis. The underlying mechanism can be explained by multiple targets of S-gluathionylation, which mediate the angiogenic effects in ischemia. Increments in the master angiogenic transcriptional factor, HIF-1α, reduction of the anti-angiogenic factor sFlt1, activation of the endoplasmic reticulum Ca2+
pump, SERCA, and inhibition of phosphatases may occur as a consequence of enhanced S-glutathionylation in ischemic tissue. In summary, inducing S-glutathionylation by inhibiting Glrx may be a therapeutic strategy to improve ischemic angiogenesis in CVD.
Reactive oxygen species (ROS) are generated mainly as result of cellular respiration, and they are maintained at low levels by antioxidant molecules and enzymes.1
It is well known that ROS are increased in cardiovascular diseases (CVD), such as myocardial infarction (MI), stroke, heart failure (HF), atherosclerosis and peripheral arterial disease.2
Increased ROS are considered a consequence of CVD, particularly as they are produced by inflammatory leukocytes, and in addition may exacerbate disease because of deleterious cellular effects caused by “oxidative stress”.3
Therefore, eliminating ROS would seem a promising strategy for treatment of CVD, particularly based on results of animal studies. However, the results of clinical trials have been far below this expectation.4
ROS are now recognized not only as cytotoxic molecules but also as cell signaling mediators.5
Little is known about the effects of ROS related to signaling in CVD. However, animal studies show that ROS contribute to improvement of CVD, including angiogenesis following ischemia.6,7
In this review, we summarize the results of antioxidants used in clinical trials and mouse CVD models, the mechanism by which ROS regulate cell signaling, and discuss how ROS influence angiogenesis. We especially focus on ROS-induced signaling by
S-glutathionylation or GSH protein adducts, which are a relatively stable oxidative post-translational protein modification.
Effects of Antioxidants on Human and Murine CVD
Levels of ROS are increased in almost every CVD. For example, ischemia reperfusion associated with MI or stroke causes a burst of ROS in mitochondria.8
Angiotensin II, and cytokines that are upregulated in HF increase ROS from NADPH oxidase.9
Macrophage-derived foam cells release ROS in atherosclerotic plaques.10
In addition, increments in ROS exacerbate CVD. For example, mouse studies show that deletion of antioxidant enzymes, including superoxide dismutase (SOD)11,12
and glutamate-cysteine ligase modifier subunit (Gclm),13
exacerbates ischemic reperfusion injury. SOD1 or SOD2 knockout (KO) mice also experience worsened stroke,14,15
and mice deficient in SOD216
or Gclm17
have impaired cardiac function in HF. Furthermore, genetic polymorphisms in antioxidant enzymes, such as SOD218
and Gclm,17,19,20
are also a risk factor for CVD. Therefore, it would be expected that treating CVD with antioxidants might be a good strategy. Actually, overexpression of antioxidant enzymes such as SOD or glutathione peroxidase (Gpx), as well as treatment with the free radical scavengers, edaravone or tempol, improves animal models of MI,21,22
ischemic reperfusion23–27
and stroke28,29
(Table). In addition, clinical studies showed that edaravone, which has been clinically used for patients in Japan, is effective in MI30
and stroke.31
In contrast to acute CVD, the effect of antioxidant therapeutics in chronic CVD has been limited. In mice, the antioxidant probucol improved HF,32
but clinical trials of antioxidant in HF have been controversial33,34
(Table). Several large clinical trials of antioxidants in the prevention of CVD were negative. These studies revealed that antioxidant vitamins C, E, and β-carotene, alone or in combination, did not reduce the risk of CVD.4
It is not clear why these studies had disappointing results. One possibility is that antioxidants have not proven universally beneficial in CVD in animal models. For example, in hindlimb ischemia models, overexpression of catalase7
or treatment with tempol6
impaired blood flow recovery. In spite of many studies that showed beneficial effects of antioxidants on MI, the antioxidant
N-acetyl cysteine decreased collateral growth after MI.35
In addition, the cardioprotective effects of ischemic preconditioning are eliminated by antioxidants.36
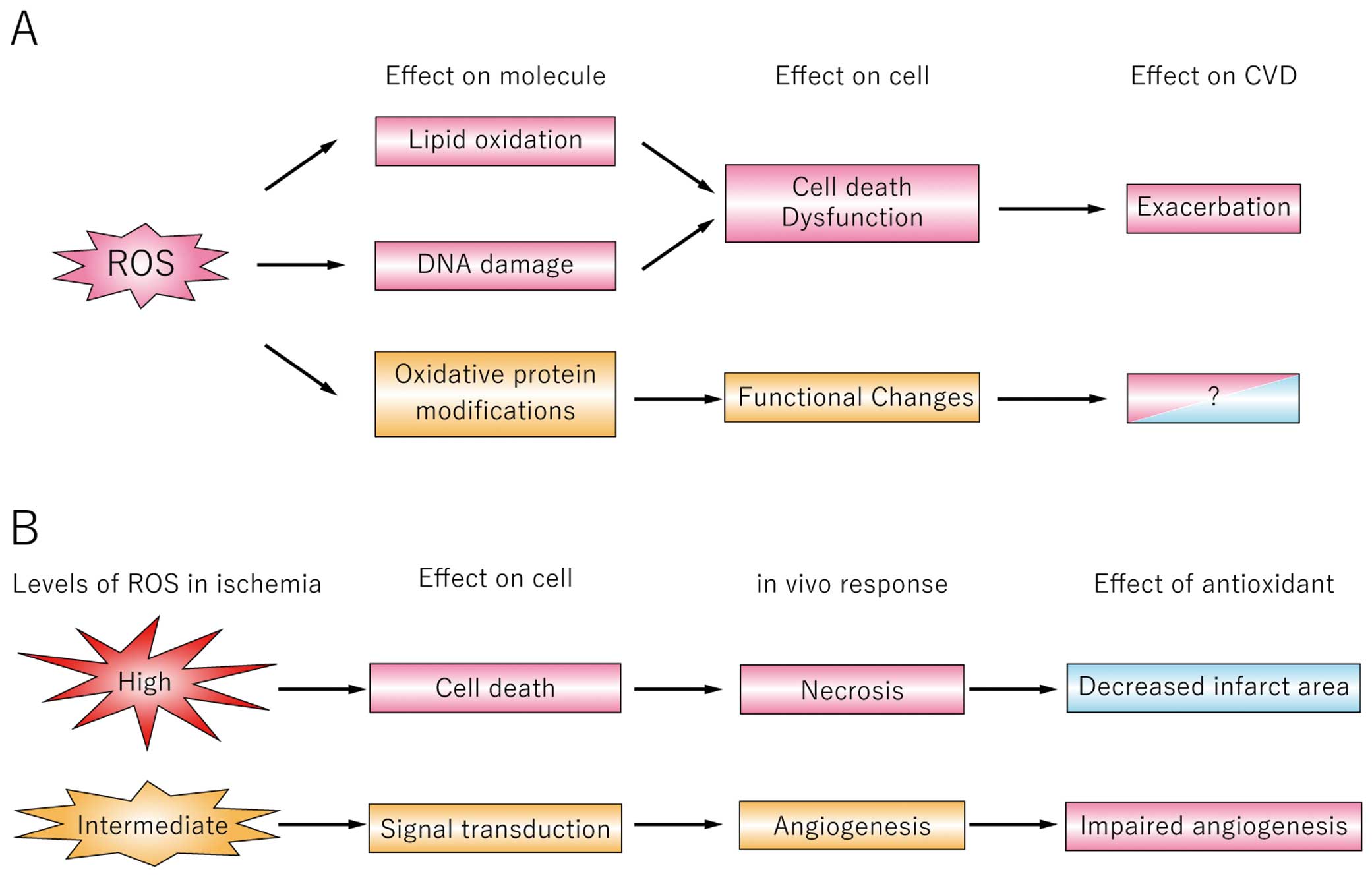
Taken together, antioxidant therapeutics may be effective in acute CVD such as MI and stroke, which are associated with high levels of ROS, but not in chronic CVDs. High levels of ROS may damage lipids and DNA by oxidation, result in cell death or dysfunction, and exacerbate CVD (Figure 1A). On the other hand, physiological levels of ROS can stimulate signaling by modifying protein function, but their role in CVD remains controversial. At the very least, ROS are likely important for angiogenic responses after ischemia because not only do antioxidants impair angiogenesis, but also overexpression of Nox4,37
which produces hydrogen peroxide, improves angiogenesis after hindlimb ischemia (Figure 1B). Thus, ROS-induced signaling, which is mediated by oxidative protein modifications, is worth considering in angiogenesis.
Table.
(A) Effect of Antioxidants or Antioxidant Enzymes in Animal Models of CVD, (B) Clinical Trials of Antioxidants in CVD
| CVD |
Exacerbate |
Improve |
| A |
| MI |
NAC (collateral growth)35 |
Gpx overexpression,21 edaravone22 |
| Ischemic reperfusion |
|
SOD1, 23 224 and Gpx25 overexpression,
edaravone,26 tempol27 |
| Stroke |
|
SOD1 overexpression,28 edaravone29 |
| HF |
|
Probucol32 |
| Hindlimb ischemia |
Catalase overexpression,7 tempol6 |
|
| Ischemic preconditioning |
SOD, N-2-mercaptopropionyl glycine36 |
|
| CVD |
Ineffective |
Improve |
| B |
| MI |
|
Edaravone30 |
| Stroke |
|
Edaravone31 |
| HF |
Vitamin E33 |
NAC (renal failure)34 |
| Prevention of CVD |
Vitamins C and E, β-carotine4 |
|
CVD, cardiovascular disease; HF, heart failure; MI, myocardial infarction; NAC, N-acetyl cysteine.
Oxidative Post-Translational Modification of Cysteine Residues
Protein oxidation results from various oxidations of amino acid residues. For example, all kinds of amino acid residues can be carbonylated and this modification is well known as a marker for protein oxidation.38
However, compared with other amino acid oxidative modifications, many studies have shown abundant cysteine modifications by ROS. Cysteine has oxidant-sensitive thiols (-SH groups) that can be modified all kinds of ROS.39
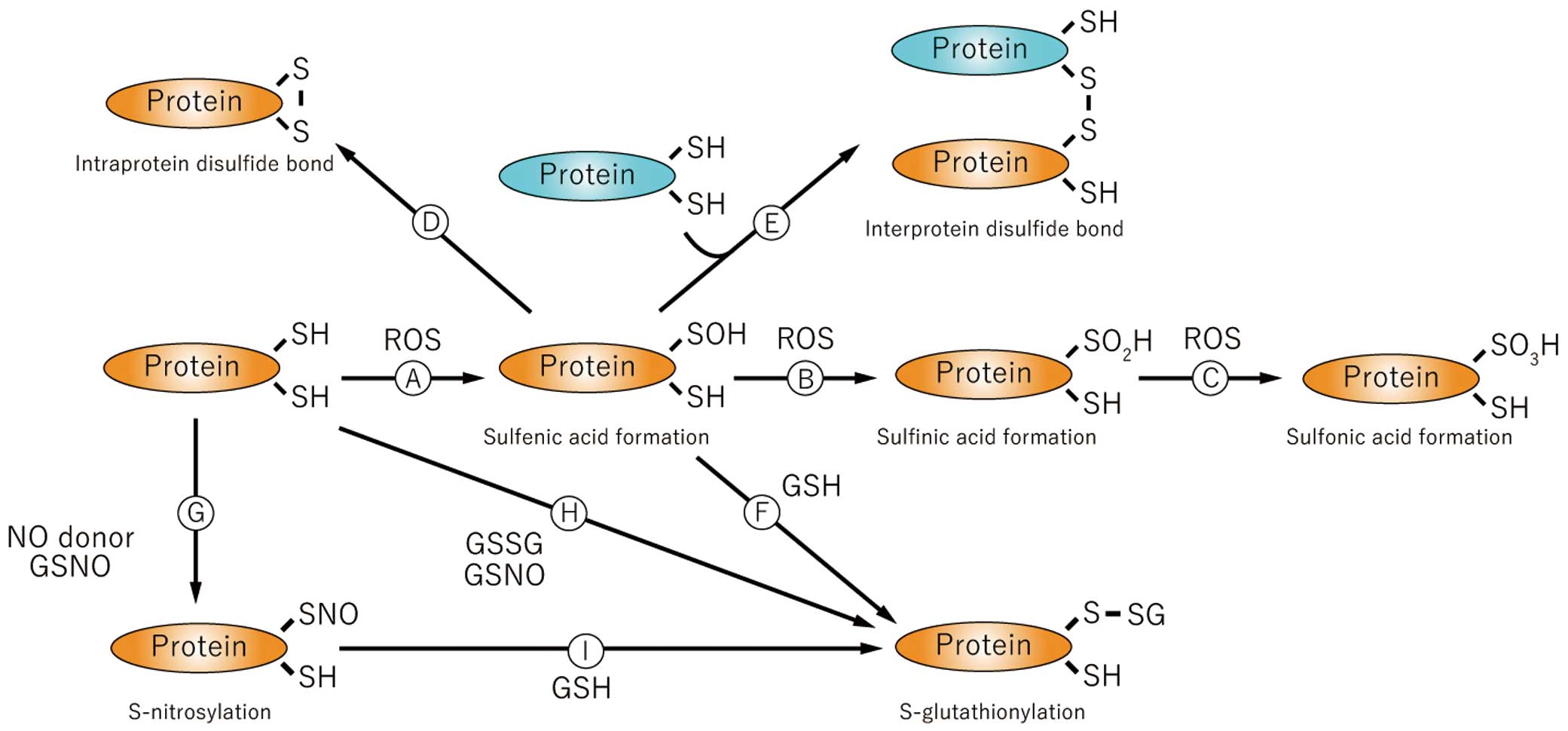
Approximately 10% of protein thiols exist as thiolate anions (-S–) because of local electrostatic forces, making them much more reactive than the uncharged thiol. These thiols in particular are easily oxidized by ROS to form sulfenic acid (Cys-SOH) (Figure 2
[A]). Because of the instability of sulfenic acid, sulfinic acid (Cys-SO2H) (Figure 2
[B]) is formed by additional oxidation. Further oxidation induces sulfonic acid (Cys-SO3H) (Figure 2
[C]). Sulfinic acid and sulfonic acid are biologically irreversible oxidative modifications, but sulfenic acid is a reducible modification or can react to form other protein modifications. Intraprotein (Figure 2
[D]) or interprotein (Figure 2
[E]) disulfide bonds may form between sulfenic acid groups. Disulfide bond formation also occurs between sulfenic acid and the most abundant cytosolic small molecule thiol, glutathione (GSH) (Figure 2
[F]), producing
S-glutathionylation. Glutathione, a tripeptide that is composed of glutamine, cysteine and glycine, can be oxidized to form the homodimer, GSSG.40
When levels of GSSG are increased under oxidant stress conditions, GSSG can form
S-glutathione adducts by thiol exchange (Figure 2
[H]). Nitric oxide (•NO), a free radical, mediates its bioactivity largely by modifying protein thiols (-SNO,
S-nitrosylation)41
(Figure 2
[G]).
S-nitrosylation is chemically unstable and may further react with GSH to form
S-glutathionylation (Figure 2
[I]). GSH may be oxidized by •NO to form
S-nitrosoglutathione (GSNO), which is relatively stable compared with •NO and also stimulates
S-nitrosylation of proteins (Figure 2
[G]) as well
S-glutathionylation (Figure 2
[H]).42
All these modifications are reported to affect protein function or signal transduction. For example, intraprotein disulfide bond formation is an important part of thioredoxin function,43
and interprotein disulfide bond formation activates ASK1.44
These protein modifications are site-specific reactions, because the reaction between 2 intra- or interprotein cysteine thiols completely depends on protein structure. Sulfenic, sulfinic, and sulfonic acids,
S-nitrosylation and
S-glutathionylation also depend on protein structure as it pertains to reactivity of cysteine residues. For example, only 1 of 14 cysteine thiols on the protein surface of SERCA exists as a thiolate anion at physiological pH, and therefore modification of its thiol represents more than 90% of the glutathione adducts as demonstrated by mutating the one cysteine to serine.45
In addition, modification of only specific cysteine residues in specific proteins results in functional changes, and it is likely that protein structure has evolved to increase the reactivity of thiols that can influence the function of a particular protein in an advantageous manner. Sulfinic and sulfonic acids, being irreversible modifications, cause protein dysfunction and degradation, whereas reversible modifications (S-sulfenylation,
S-nitrosylation and
S-glutathionylation) are considered to play an important role in the induction of ROS-related signaling. All these modifications give negative charge to cysteine residue, and may change protein function.
S-nitrosylation (-SNO) and sulfenic acid (-SOH) react with intracellular GSH to form the much more stable
S-glutathionylation (-SSG).46
S-glutathionylation is greatly increased in ischemic muscles for at least several days after hindlimb ischemia,47
but whether other modifications occur physiologically under the same conditions is unknown. Therefore, particularly as pertains to more chronic influences of ROS,
S-glutathionylation might play an important role in angiogenesis after ischemia.
Glutaredoxin and Its Role in Ischemic Revascularization
Removal of GSH protein adducts is catalyzed by a small cytosolic enzyme, glutaredoxin-1 (Glrx). Glrx, also called thioltransferase, specifically catalyzes the removal of GSH adducts from
S-glutathionylated proteins using one of its own thiols on cysteine 22 or 25.48
After GSH is removed from a protein, Glrx requires GSH to be returned to an active reduced form again. The GSH that is oxidized to GSSG in the process needs to be reduced by glutathione reductase, which requires NADPH (Figure 3A). Thus, Glrx activity depends on GSH, glutathione reductase, and NADPH levels in cells. Glrx generates dynamic redox signaling by removing GSH protein adducts and thus may control pathophysiological phenotypes. Upregulation of Glrx inhibits VEGF-induced endothelial cell migration49
and activates NF-κB signaling.47
Deletion of endogenous Glrx in mice attenuates cardiovascular hypertrophy induced by angiotensin II in association with lower ROS production in the aorta.50
Therefore, the role of Glrx in controlling GSH adducts goes beyond antioxidant effects. Glrx expression is increased in atherosclerotic human coronary artery51
and diabetic rat retina.52
Glrx itself is induced by NF-κB activation53
and is therefore upregulated in association with inflammation. However, the pathophysiological role of Glrx in vivo still needs to be explored.
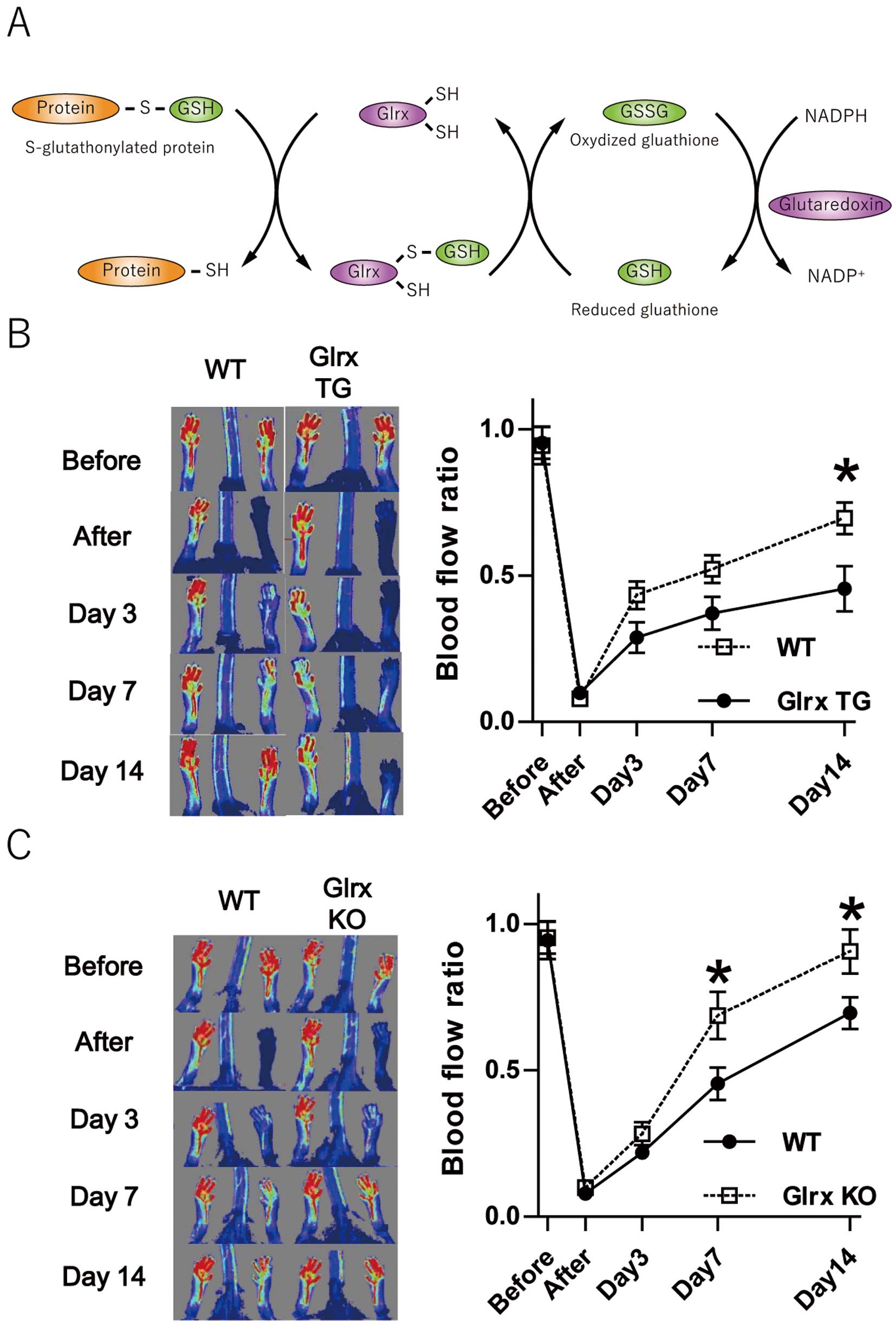
Just as anti-angiogenic effects are shown by Glrx overexpression in vitro,49
Glrx overexpressing transgenic (TG) mice show poor blood flow recovery in hindlimb ischemia compared with control wild-type mice47
(Figure 3B). The impaired blood flow is associated with poor motor function and lower capillary density in muscle, reflecting poorer angiogenesis. In contrast, deletion of Glrx in the KO mice improves revascularization after hindlimb ischemia54
(Figure 3C). Therefore, these data indicate that endogenous Glrx has an anti-angiogenic role. In other words, oxidant-induced S-glutathionylation (GSH adducts) on protein(s) in ischemic muscles facilitate angiogenic responses and promote ischemic revascularization. There is a report55
indicating that Glrx overexpression enhances neovascularization in a mouse MI model; however, they evaluated vascularization only by α-smooth muscle actin staining in hearts, and the results are not sufficient to conclude Glrx enhances neovascularization. Also, they showed anti-apoptotic effects in cardiomyocytes, which most likely explain the better cardiac function observed in Glrx TG mice.
Role of S-Glutathionylation in Angiogenesis
Gain and loss of function of Glrx in vivo shows that
S-glutathionylation stimulates angiogenic signaling after ischemia (Figure 4). Vascular endothelial growth factor A (VEGFa), the central angiogenic growth factor, plays an important role in angiogenesis and ischemic revascularization.
S-glutathionylation regulates VEGFa production and signaling through effects on multiple proteins. First,
S-
glutathionylation of Cys520 of HIF1α stabilizes the protein and increases VEGFa expression.54
Second,
S-glutathionylation of the NF-κB components p65,47
p50,56
and Inhibitor of κ B kinase (IKK)57
inhibits NF-κB activation. Soluble Flt (sFlt)-1, which binds to VEGFa as a decoy receptor and inhibits VEGFa signaling, is regulated by NF-κB and is anti-angiogenic. Decreased
S-glutathionylation of p65 caused by Glrx overexpression results in hyper-activation of NF-κB and increased sFlt1,47
indicating that
S-glutathionylation has a role in suppressing anti-angiogenic sFlt1 production. Therefore,
S-glutathionylation promotes VEGFa expression and activation of its signaling receptor (VEGFR2) by decreasing this inhibitory factor.
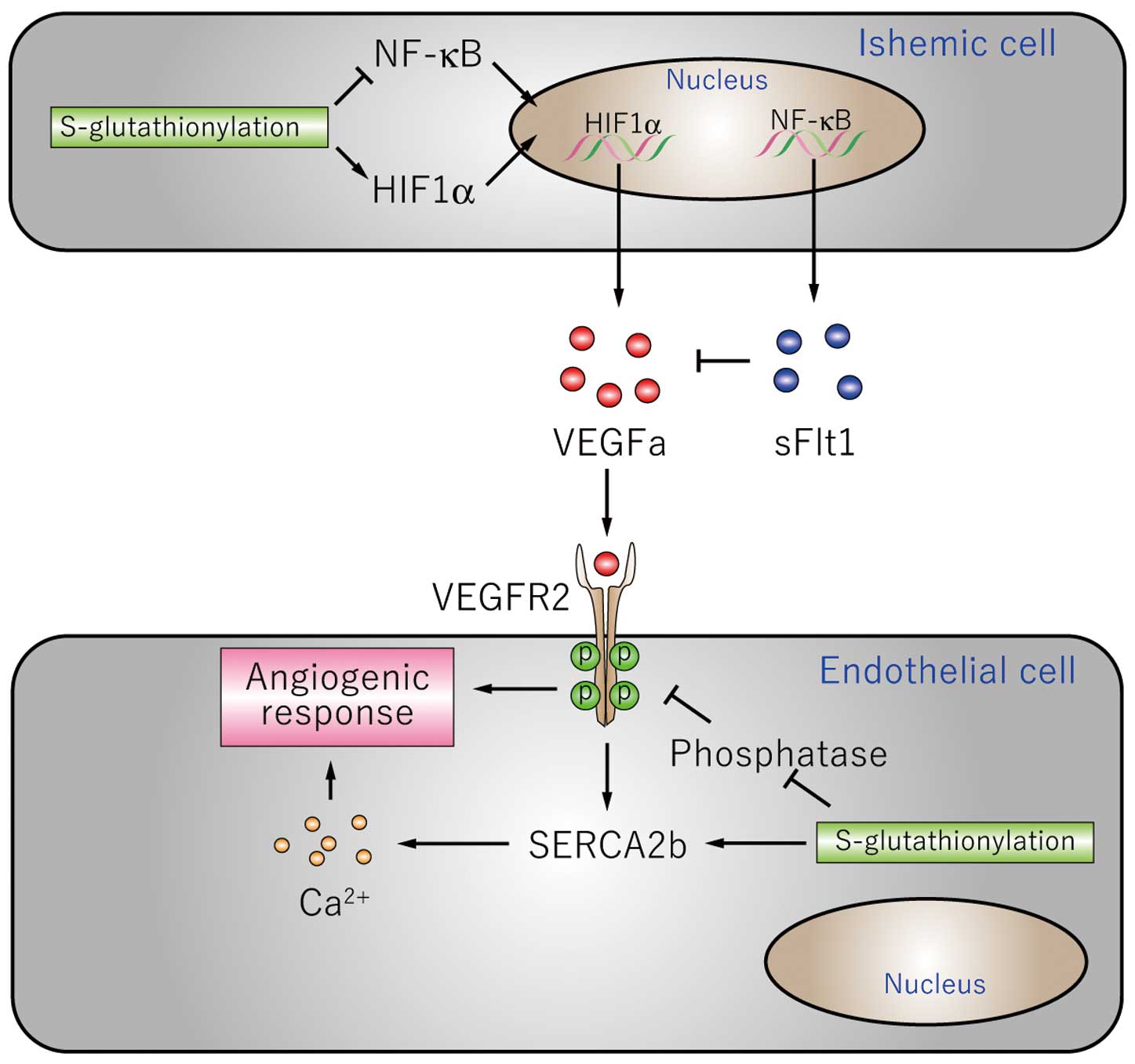
In addition, downstream of VEGF receptor activation,
S-glutathionylation regulates VEGFa-dependent angiogenic responses in endothelial cells. VEGFa-dependent Ca2+
influx is critical for angiogenic responses in endothelial cells. The sarco/endoplasmic reticulum Ca2+
ATPase (SERCA) 2b is activated by S-glutathionylation of Cys674.45
GSH adducts on Cys674 are required for VEGF-induced increased SERCA activity (ER Ca2+
uptake), increased cytosolic Ca2+
influx, and endothelial cell migration.49
In addition, in vivo S-glutathionylation of SERCA2b C674 is essential for normal angiogenesis
after hindlimb ischemia in mice.58
Those studies indicate that
S-glutathionylation of SERCA C674 critically regulates endothelial angiogenic responses.
As another example, phosphatases, such as protein tyrosine phosphatase-1B (PTP1B) and protein phosphatase-A2 (PP2A), normally suppress VEGFa signaling and are inhibited by oxidative protein modifications.59
GSH adducts on Cys215 of PTP1B inhibit phosphatase activity.60
In endothelial cells, PTP1B inhibits tyrosine phosphorylation of the VEGF receptor, VEGFR2,61
and deletion of PTP1B in mice increases angiogenesis after MI.62
Therefore, the inhibition of phosphatases by GSH adducts may enhance tyrosine phosphorylation of VEGFR2, thereby enhancing the activity of focal adhesion kinase, a key mediator of endothelial cell migration, and angiogenesis.
In ischemic conditions, various other proteins are
S-glutathionylated. It is reported that some pro-angiogenic proteins, such as eNOS63
and sirtuin-1,64
are inhibited by
S-glutathionylation, and therefore might be expected to inhibit their important role in angiogenesis. This apparent contradiction could exist for several reasons, including the possibility that the proteins are protected from
S-glutathionylation during ischemia because of their subcellular localization (eg, calveoli and nucleus, respectively) or the effects are masked by other angiogenic factors. However, in vivo studies of Glrx KO and overexpression indicate that overall enhancement of
S-glutathionylation by inhibiting Glrx promotes angiogenenic responses during ischemia.
Summary
ROS are increased in CVD and considered a deleterious consequence of the diseases. However, antioxidant therapeutics in humans do not prevent CVD. This might be explained by a beneficial effect of ROS on CVD, especially in ischemic angiogenesis. Although high levels of ROS cause cell damage and death, another aspect of ROS is to directly modify protein thiols and change protein function. S-glutathionylation, a relatively stable oxidative cysteine modification by GSH, increases during hindlimb ischemia and apparently stimulates angiogenic signaling. Studies of gain and loss of function of Glrx, which specifically removes S-glutathionylation from proteins, reveal that S-glutathionylation, on balance, stimulates angiogenesis after hindlimb ischemia. S-glutathionylation increases active VEGFa by stabilizing HIF-1α and inhibiting the anti-angiogenic factor, sFlt1, as well as enhancing VEGFa signaling in endothelial cells through inhibition of PTPs and activation of SERCA2. Therefore, S-glutathionylation and its regulator Glrx may be good targets for treatment to improve ischemic angiogenesis in CVD.
Acknowledgments
The work summarized in this review and writing of the article were supported by funding from the National Institutes of Health grants RO1HL31607 and R37HL104017 (R.A.C.), RO1HL133013 (R.M), Sumitomo Life Welfare and Culture Foundation, as well as Mochida Memorial Foundation for Medical and Pharmaceutical Research (Y.W.).
Disclosure
R.A.C. is the recipient of grants from the Servier Pharmaceutical company not on the subject of this review.
References
- 1.
Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 2001; 31: 1287–1312.
- 2.
Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 2005; 115: 500–508.
- 3.
Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury part I: Basic mechanisms and in vivo monitoring of ROS. Circulation 2003; 108: 1912–1916.
- 4.
Kris-Etherton PM, Lichtenstein AH, Howard BV, Steinberg D, Witztum JL; Nutrition Committee of the American Heart Association Council on Nutrition,
Physical Activity, and Metabolism. Antioxidant vitamin supplements and cardiovascular disease. Circulation 2004; 110: 637–641.
- 5.
D’Autréaux B, Toledano MB. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat Rev Mol Cell Biol 2007; 8: 813–824.
- 6.
Kim HW, Lin A, Guldberg RE, Ushio-Fukai M, Fukai T. Essential role of extracellular SOD in reparative neovascularization induced by hindlimb ischemia. Circ Res 2007; 101: 409–419.
- 7.
Urao N, Sudhahar V, Kim SJ, Chen GF, McKinney RD, Kojda G, et al. Critical role of endothelial hydrogen peroxide in post-ischemic neovascularization. PLoS One 2013; 8: e57618, doi:10.1371/journal.pone.0057618.
- 8.
Di Lisa F, Bernardi P. Mitochondria and ischemia-reperfusion injury of the heart: Fixing a hole. Cardiovasc Res 2006; 70: 191–199.
- 9.
Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase role in cardiovascular biology and disease. Circ Res 2000; 86: 494–501.
- 10.
Rajagopalan S, Meng XP, Ramasamy S, Harrison DG, Galis ZS. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro: Implications for atherosclerotic plaque stability. J Clin Invest 1996; 98: 2572.
- 11.
Yoshida T, Maulik N, Engelman RM, Ho YS, Das DK. Targeted disruption of the mouse Sod I gene makes the hearts vulnerable to ischemic reperfusion injury. Circ Res 2000; 86: 264–269.
- 12.
Kim GW, Kondo T, Noshita N, Chan PH. Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice: Implications for the production and role of superoxide radicals. Stroke 2002; 33: 809–815.
- 13.
Kobayashi T, Watanabe Y, Saito Y, Fujioka D, Nakamura T, Obata J, et al. Mice lacking the glutamate-cysteine ligase modifier subunit are susceptible to myocardial ischaemia-reperfusion injury. Cardiovasc Res 2010; 85: 785–795.
- 14.
Kondo T, Reaume AG, Huang TT, Murakami K, Carlson E, Chen S, et al. Edema formation exacerbates neurological and histological outcomes after focal cerebral ischemia in CuZn-superoxide dismutase gene knockout mutant mice. Acta Neurochir Suppl 1997; 70: 62–64.
- 15.
Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, et al. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci 1998; 18: 205–213.
- 16.
Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, et al. Oxidative stress causes heart failure with impaired mitochondrial respiration. J Biol Chem 2006; 281: 33789–33801.
- 17.
Watanabe Y, Watanabe K, Kobayashi T, Saito Y, Fujioka D, Nakamura T, et al. Chronic depletion of glutathione exacerbates ventricular remodelling and dysfunction in the pressure-overloaded heart. Cardiovasc Res 2013; 97: 282–292.
- 18.
Hiroi S, Harada H, Nishi H, Satoh M, Nagai R, Kimura A. Polymorphisms in the SOD2 and HLA-DRB1 genes are associated with nonfamilial idiopathic dilated cardiomyopathy in Japanese. Biochem Biophys Res Commun 1999; 261: 332–339.
- 19.
Nakamura S, Kugiyama K, Sugiyama S, Miyamoto S, Koide S, Fukushima H, et al. Polymorphism in the 5’-flanking region of human glutamate-cysteine ligase modifier subunit gene is associated with myocardial infarction. Circulation 2002; 105: 2968–2973.
- 20.
Nakamura S, Sugiyama S, Fujioka D, Kawabata K, Ogawa H, Kugiyama K. Polymorphism in glutamate-cysteine ligase modifier subunit gene is associated with impairment of nitric oxide-mediated coronary vasomotor function. Circulation 2003; 108: 1425–1427.
- 21.
Shiomi T, Tsutsui H, Matsusaka H, Murakami K, Hayashidani S, Ikeuchi M, et al. Overexpression of glutathione peroxidase prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2004; 109: 544–549.
- 22.
Onogi H, Minatoguchi S, Chen XH, Bao N, Kobayashi H, Misao Y, et al. Edaravone reduces myocardial infarct size and improves cardiac function and remodelling in rabbits. Clin Exp Pharmacol Physiol 2006; 33: 1035–1041.
- 23.
Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, et al. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke 1994; 25: 165–170.
- 24.
Chen Z, Siu B, Ho YS, Vincent R, Chua CC, Hamdy RC, et al. Overexpression of MnSOD protects against myocardial ischemia/reperfusion injury in transgenic mice. J Mol Cell Cardiol 1998; 30: 2281–2289.
- 25.
Weisbrot-Lefkowitz M, Reuhl K, Perry B, Chan PH, Inouye M, Mirochnitchenko O. Overexpression of human glutathione peroxidase protects transgenic mice against focal cerebral ischemia/reperfusion damage. Brain Res Mol Brain Res 1998; 53: 333–338.
- 26.
Yagi H, Horinaka S, Matsuoka H. Edaravone prevented deteriorated cardiac function after myocardial ischemia-reperfusion via inhibiting lipid peroxidation in rat. J Cardiovasc Pharmacol 2005; 46: 46–51.
- 27.
McDonald MC, Zacharowski K, Bowes J, Cuzzocrea S, Thiemermann C. Tempol reduces infarct size in rodent models of regional myocardial ischemia and reperfusion. Free Radic Biol Med 1999; 27: 493–503.
- 28.
Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci USA 1991; 88: 11158–11162.
- 29.
Zhang N, Komine-Kobayashi M, Tanaka R, Liu M, Mizuno Y, Urabe T. Edaravone reduces early accumulation of oxidative products and sequential inflammatory responses after transient focal ischemia in mice brain. Stroke 2005; 36: 2220–2225.
- 30.
Tsujita K, Shimomura H, Kawano H, Hokamaki J, Fukuda M, Yamashita T, et al. Effects of edaravone on reperfusion injury in patients with acute myocardial infarction. Am J Cardiol 2004; 94: 481–484.
- 31.
Edaravone Acute Infarction Study Group. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction: Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc Dis 2003; 15: 222.
- 32.
Sia YT, Lapointe N, Parker TG, Tsoporis JN, Deschepper CF, Calderone A, et al. Beneficial effects of long-term use of the antioxidant probucol in heart failure in the rat. Circulation 2002; 105: 2549–2555.
- 33.
Keith ME, Jeejeebhoy KN, Langer A, Kurian R, Barr A, O’Kelly B, et al. A controlled clinical trial of vitamin E supplementation in patients with congestive heart failure. Am J Clin Nutr 2001; 73: 219–224.
- 34.
Tepel M, Van Der Giet M, Statz M, Jankowski J, Zidek W. The antioxidant acetylcysteine reduces cardiovascular events in patients with End-stage renal failure a randomized, controlled trial. Circulation 2003; 107: 992–995.
- 35.
Gu W, Weihrauch D, Tanaka K, Tessmer JP, Pagel PS, Kersten JR, et al. Reactive oxygen species are critical mediators of coronary collateral development in a canine model. Am J Physiol Heart Circ Physiol 2003; 285: H1582–H1589.
- 36.
Tanaka M, Fujiwara H, Yamasaki K, Sasayama S. Superoxide dismutase and N-2-mercaptopropionyl glycine attenuate infarct size limitation effect of ischaemic preconditioning in the rabbit. Cardiovasc Res 1994; 28: 980–986.
- 37.
Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, et al. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011; 124: 731–740.
- 38.
Shacter E. Quantification and significance of protein oxidation in biological samples. Drug Metab Rev 2000; 32: 307–326.
- 39.
Finkel T. Signal transduction by reactive oxygen species. J Cell Biol 2011; 194: 7–15.
- 40.
Meister A, Anderson ME. Glutathione. Annu Rev Biochem 1983; 52: 711–760.
- 41.
Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: Purview and parameters. Nat Rev Mol Cell Biol 2005; 6: 150–166.
- 42.
Ji Y, Akerboom TP, Sies H, Thomas JA. S-nitrosylation and S-glutathiolation of protein sulfhydryls bys-nitroso glutathione. Arch Biochem Biophys 1999; 362: 67–78.
- 43.
Holmgren A. Thioredoxin. Annu Rev Biochem 1985; 54: 237–271.
- 44.
Jarvis RM, Hughes SM, Ledgerwood EC. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic Biol Med 2012; 53: 1522–1530.
- 45.
Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, et al. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med 2004; 10: 1200–1207.
- 46.
Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem 2000; 267: 4928–4944.
- 47.
Murdoch CE, Shuler M, Haeussler DJ, Kikuchi R, Bearelly P, Han J, et al. Glutaredoxin-1 up-regulation induces soluble vascular endothelial growth factor receptor 1, attenuating post-ischemia limb revascularization. J Biol Chem 2014; 289: 8633–8644.
- 48.
Shelton MD, Mieyal JJ. Regulation by reversible S-glutathionylation: Molecular targets implicated in inflammatory diseases. Mol Cell 2008; 25: 332.
- 49.
Evangelista AM, Thompson MD, Weisbrod RM, Pimental DR, Tong X, Bolotina VM, et al. Redox regulation of SERCA2 is required for vascular endothelial growth factor-induced signaling and endothelial cell migration. Antioxid Redox Signal 2012; 17: 1099–1108.
- 50.
Bachschmid MM, Xu S, Maitland-Toolan KA, Ho YS, Cohen RA, Matsui R. Attenuated cardiovascular hypertrophy and oxidant generation in response to angiotensin II infusion in glutaredoxin-1 knockout mice. Free Radic Biol Med 2010; 49: 1221–1229.
- 51.
Okuda M, Inoue N, Azumi H, Seno T, Sumi Y, Hirata K, et al. Expression of glutaredoxin in human coronary arteries its potential role in antioxidant protection against atherosclerosis. Arterioscler Thromb Vasc Biol 2001; 21: 1483–1487.
- 52.
Shelton MD, Kern TS, Mieyal JJ. Glutaredoxin regulates nuclear factor kappa-B and intercellular adhesion molecule in Muller cells: Model of diabetic retinopathy. J Biol Chem 2007; 282: 12467–12474.
- 53.
Ather JL, Hodgkins SR, Janssen-Heininger YM, Poynter ME. Airway epithelial NF-κB activation promotes allergic sensitization to an innocuous inhaled antigen. Am J Respir Cell Mol Biol 2011; 44: 631–638.
- 54.
Watanabe Y, Murdoch CE, Sano S, Ido Y, Bachschmid MM, Cohen RA, et al. Glutathione adducts induced by ischemia and deletion of glutaredoxin-1 stabilized HIF-1 α and improve limb revascularization. Proc Natl Acad Sci USA
2016 (in press).
- 55.
Adluri RS, Thirunavukkarasu M, Zhan L, Dunna NR, Akita Y, Selvaraju V, et al. Glutaredoxin-1 overexpression enhances neovascularization and diminishes ventricular remodeling in chronic myocardial infarction. PLoS One 2012; 7: e34790, doi:10.1371/journal.pone.0034790.
- 56.
Pineda-Molina E, Klatt P, Vázquez J, Marina A, García de Lacoba M, Pérez-Sala D, et al. Glutathionylation of the p50 subunit of NF-κB: A mechanism for redox-induced inhibition of DNA binding. Biochemistry 2001; 40: 14134–14142.
- 57.
Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, et al. Dynamic redox control of NF-κB through glutaredoxin-regulated S-glutathionylation of inhibitory κB kinase β. Proc Natl Acad Sci USA 2006; 103: 13086–13091.
- 58.
Thompson MD, Mei Y, Weisbrod RM, Silver M, Shukla PC, Bolotina VM, et al. Glutathione adducts on sarcoplasmic/endoplasmic reticulum Ca2+ ATPase Cys-674 regulate endothelial cell calcium stores and angiogenic function as well as promote ischemic blood flow recovery. J Biol Chem 2014; 289: 19907–19916.
- 59.
Rao R, Clayton L. Regulation of protein phosphatase 2A by hydrogen peroxide and glutathionylation. Biochem Biophys Res Commun 2002; 293: 610–616.
- 60.
Barrett WC, DeGnore JP, König S, Fales HM, Keng YF, Zhang ZY, et al. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry 1999; 38: 6699–6705.
- 61.
Nakamura Y, Patrushev N, Inomata H, Mehta D, Urao N, Kim HW, et al. Role of protein tyrosine phosphatase 1B in vascular endothelial growth factor signaling and cell-cell adhesions in endothelial cells. Circ Res 2008; 102: 1182–1191.
- 62.
Besnier M, Galaup A, Nicol L, Henry JP, Coquerel D, Gueret A, et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. FASEB J 2014; 28: 3351–3361.
- 63.
Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MH, et al. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 2010; 468: 1115–1118.
- 64.
Zee RS, Yoo CB, Pimentel DR, Perlman DH, Burgoyne JR, Hou X, et al. Redox regulation of sirtuin-1 by S-glutathiolation. Antioxid Redox Signal 2010; 13: 1023–1032.