Review
Synthesis of Heterocyclic Compounds through Palladium-Catalyzed C–H Cyclization Processes
2013 Volume 61 Issue 10 Pages 987-996
Details
2013 Volume 61 Issue 10 Pages 987-996
Herein, we describe our development of synthetic methods for heterocyclic compounds based on the palladium-catalyzed carbon–hydrogen bond (C–H) functionalization/intramolecular carbon–heteroatom (nitrogen or sulfur) bond formation process. By this C–H cyclization method, we efficiently prepared various N-heterocycles, including indazoles, indoles, and 2-quinolinones, as well as S-heterocycles such as benzothiazoles and benzo[b]thiophenes. Yields are typically good to high and good functional-group tolerance is observed for each process, thereby indicating that the method provides a novel, highly applicable synthetic route to the abovementioned biologically important heterocyclic frameworks. As an application of this approach, an auto-tandem-type, one-pot process involving the oxidative Heck reaction and subsequent C–H cyclization using cinnamamides and arylboronic acids as starting materials in the presence of a palladium catalyst was also developed for the rapid construction of the 2-quinolinone nucleus.
In recent years, substantial progress has been made in the development of new transformations via the transition-metal-catalyzed functionalization of unreactive C–H bonds, which can provide more efficient and straightforward alternatives to conventional cross-coupling methods. Palladium-based systems are among the most frequently employed catalysts for C–H functionalization, exhibiting versatile catalytic activities with various reaction patterns.1–10) In this research area, however, most investigations have focused on carbon–carbon (C–C), carbon–oxygen (C–O), and carbon–halogen (C–X) bond formation, whereas less attention has been paid to carbon–nitrogen (C–N)11) and carbon–sulfur (C–S) bond formation.12) Our group has been working on the palladium-catalyzed formation of C–N and C–S bonds via C–H functionalization, specifically applying it to intramolecular processes, which would provide novel routes to N- and S-heterocycles.7,13,14) The working hypothesis at our starting point is shown in Chart 1. The treatment of compound 1, which possesses a heteroatom-based nucleophilic part (YH, Y=NR or S, which also functions as a directing group), with a palladium(II) catalyst forms palladium complex 3 via 2. The formation of palladacycle 4, which could result from the C–H functionalization of 3, is followed by reductive elimination, which produces heterocyclic compound 5 along with palladium(0). The latter can be oxidized into palladium(II) by the use of a reoxidant, thereby rendering the process catalytic. In this review, we present a comprehensive overview of our studies on the construction of heterocycles by palladium-catalyzed C–H functionalization.

Nitrogen is an essential atom in nature, existing in a wide range of naturally occurring compounds, such as alkaloids and amino acids, as well as in designed medicinal agents. Therefore, the development of methods to introduce a nitrogen–atom-containing moiety into certain molecules, especially via C–N bond formation, has been a research area of considerable importance. As a transition-metal-catalyzed process, the palladium-catalyzed amination of aryl/vinyl (pseudo)halides (the so-called Buchwald–Hartwig amination) has tremendously evolved over the past two decades.15–17) Considerable effort has been devoted to obtaining efficient catalytic systems, mainly via the design of appropriate ligands. However, despite its usefulness, the method requires a halogen atom in starting electrophiles, which sometimes limits its practical use in organic synthesis. However, C–N bond formation via C–H functionalization (C–H amination) provides a complementary approach to the Buchwald–Hartwig amination process, thereby enabling the realization of more step- and atom-economical organic syntheses.9,18–21) During the course of our research program aimed at the efficient construction of heterocyclic compounds, we envisioned the use of intramolecular C–H amination for the development of novel, efficient synthetic routes to various N-heterocycles.
2.1. Indazole Synthesis22)Indazole derivatives, bioisosteres of indoles, constitute an important class of N-heterocycles in the medicinal area.23) We anticipated that benzophenone tosylhydrazone 6a would undergo intramolecular C–H amination with the aid of a palladium catalyst to afford 3-substituted indazoles 7a.24) Through the systematic screenings of a range of palladium sources, reoxidants, and solvents, we found that the desired cyclization did occur in dimethyl sulfoxide (DMSO) at 50°C in the presence of a catalytic amount of Pd(OAc)2 along with Cu(OAc)2 as a reoxidant. Interestingly, the addition of some Ag salts, especially AgOCOCF3, significantly enhanced the process (Chart 2). The use of Cu(OAc)2 or AgOCOCF3 alone as a reoxidant led to a considerable decrease in the yields.

Our examination of the substrate scope of the process revealed that various functional groups, such as electron-withdrawing –NO2, –CN, or –CO2Et and electron-donating –OMe, as well as halogen atoms such as –Br and –Cl, are well tolerated during the process. More importantly, our evaluation of the reactivity of electronically unsymmetrical substrates 6b–d revealed that the electron-rich arenes are more reactive, providing 7b–d as a sole product; this result suggests that C–H cleavage might proceed via an electrophilic palladation pathway (Chart 3).

The previously mentioned catalytic C–H amination approach was successfully applied to the cyclization of enamide 8, thereby providing a new route to a 3-substituted indole framework that exists in numerous biologically active compounds26–29) (Chart 4). The catalytic system that consisted of 10 mol % Pd(OAc)2/100 mol % Cu(OAc)2 proved optimal, and the reaction-accelerating effect of a silver salt, as mentioned in the previous section, was not observed in the indole synthesis.

2-Quinolinones, which are also subunits of a range of natural products31,32) and pharmaceuticals,33–37) can be prepared via palladium-catalyzed intramolecular C–H amidation. Specifically, the treatment of N-tosyl-3,3-diphenylacrylamide (10a) with a catalytic amount of PdCl2 along with a reoxidant such as 100 mol % Cu(OAc)2 in DMSO afforded the cyclized product (Chart 5). Under the conditions, the detosylation of 2-quinolinone unexpectedly occurred during the course of the reaction, thereby generating 4-phenyl-2-quinolinone (11a) in a fairly good yield.38) Importantly, the use of an O2 atmosphere considerably improved the yield and reduced the amount of Cu(OAc)2 to 30 mol %. Moreover, the catalytic activity of the system was also observed in the reaction conducted under an air atmosphere, which is notable from a practical viewpoint.

Reactions using several symmetrically substituted 3,3-diarylacrylamides 10b–h resulted in the successful formation of the corresponding 2-quinolinones 11b–h (Table 1).
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/T1.png%22>');%0A%09%09%09newWindow.document.close();%0A%09%09) | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | Yield (%) | ||
| 1 |  | 10b (X=F) |  | 11b (X=F) | 79 |
| 2 | 10c (X=Br) | 11c (X=Br) | 87 | ||
| 3 | 10d (X=Cl) | 11d (X=Cl) | 61 | ||
| 4 |  | 10e |  | 11e | 38 |
| 5 |  | 10f |  | 11f | 85 |
| 6a) |  | 10g |  | 11g | 86 |
| 7a) |  | 10h |  | 11h | 98 |
a) 140°C.
Although extensive mechanistic studies have yet to be conducted, experiments with isotopically labeled substrates revealed primary kinetic isotope effects (kH/kD) of 3.5 (intramolecular) and 2.7 (intermolecular), which indicate that the cleavage of the C–H bond is involved in the rate-determining step (Chart 6).

The rapid construction of structurally complex molecules from simple materials by multiple bond-forming reactions in a single step—the so-called tandem process—can be a powerful tool for synthetic organic chemists. In addition to their efficiency in organic synthesis, tandem processes reduce the number of steps in the synthesis of certain molecules; therefore, such processes are attractive from the viewpoint of the development of environmentally benign and economical synthetic methods. To develop a more efficient approach to the synthesis of heterocycles via catalytic C–H functionalization, we envisioned a tandem process consisting of the oxidative Heck reaction of cinnamamides 12 with arylboron compounds (Ar′–“B”) followed by the intramolecular C–H amidation reaction; both of these reactions can be catalyzed by palladium(II), thereby resulting in a facile and novel route to various substituted 4-aryl-2-quinolinone derivatives40) (13, Chart 7).

To probe the viability of the anticipated auto-tandem process, extensive screenings of reaction conditions were carried out using N-methoxycinnamamide (12a). Fortunately, we found that the use of phenylboronic acid (13a) as a coupling partner along with a catalytic combination of 10 mol % Pd(OAc)2/1,10-phenanthroline, 200 mol % Cu(TFA)2·nH2O, and 100 mol % Ag2O successfully effected the desired process in AcOH, delivering the cyclized compound. In this case, demethoxylation unexpectedly occurred during the reaction, resulting in the formation of N-free 2-quinolinone 11a in fairly good yields (Table 2, entries 1 and 2). Further optimization studies led to another interesting finding: the use of increased amounts of Ag2O (e.g., 300 mol % or more) completely inhibited demethoxylation and resulted in the formation of 2-quinolinone 14aa (entries 3–6). The best result was obtained when 800 mol % of Ag2O was employed, affording 14aa in high yields (entries 5 and 6). During the process, the use of a large excess of reoxidants may inhibit the undesired N–O bond cleavage in which palladium and/or copper salt(s) involve.41–43)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/T2.png%22>');%0A%09%09%09newWindow.document.close();%0A%09%09) | ||||
|---|---|---|---|---|
| Entry | Cu(TFA)·nH2O (mol%) | Ag2O (mol%) | Yield (%) | |
| 11a | 14aa | |||
| 1 | 200 | 100 | 60 | 0 |
| 2 | 100 | 100 | 56 | 0 |
| 3 | 100 | 300 | 0 | 52 |
| 4 | 100 | 400 | 0 | 64 |
| 5 | 100 | 800 | 0 | 76 |
| 6a) | 100 | 800 | 0 | 81 |
a) The reaction mixture was first stirred at 100°C for 1 h and then stirred at 120°C for 10 h.
The results of the tandem process affording 2-quinolinones via the formation of symmetrical 3,3-diarylacrylamides are summarized in Table 3. Reactions of substrates with a methyl group or a halogen atom at the para-position of the benzene rings proceeded efficiently (entries 1–3), whereas that of cinnamamide 12e, which possesses a methoxy group at the same position, resulted in the formation of the desired 2-quinolinone 14ee in only 12% yield, along with the recovery of the starting 12e in 85% yield (entry 4). In the case of the reaction of 12f, which possesses a methyl group at the meta-position of the benzene ring, C–H cyclization occurred at the less-hindered site, exclusively affording 2-quinolinone 14ff (entry 5). In contrast, the introduction of a methyl group at the ortho-position of the benzene ring did not result in the formation of the coupling or cyclized product (entry 6).
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/T3.png%22>');%0A%09%09%09newWindow.document.close();%0A%09%09) | |||||
|---|---|---|---|---|---|
| Entry | Substrate (12) | 13 | Conditionsa) | Product (14) | Yield (%) |
| 1 |  | 13b R=4-Me | A (t1=16) |  | 76 |
| 2 |  | 13c R=4-F | B (t1=3, t2=43) |  | 59 |
| 3 |  | 13d R=4-Cl | B (t1=3, t2=10) |  | 61 |
| 4 |  | 13e R=4-OMe | A (t1=12) |  | 12b) |
| 5 |  | 13f R=3-Me | A (t1=12) |  | 68 |
| 6 |  | 13g R=2-Me | A (t1=13) |  | 0c) |
a) Conditions A: performed at 100°C for t1 h. Conditions B: performed at 100°C for t1 h and then 120°C for t2 h. b) 85% of 12e was recovered. c) 34% of 12g was recovered.
Reactions via the formation of unsymmetrical 3,3-diarylacrylamides were also examined (Chart 8). For example, cinnamamide 12a reacted with boronic acid 13h under optimized conditions to successfully afford 2-quinolinone 14ah as the sole product (eq. 1). No regioisomer was identified, which suggests that the alkene isomerization of the Heck product formed in situ did not occur during the process. The same result applies to the reaction of cinnamamide 12h with 13d (eq. 2). However, when cinnamamide 12e, which possesses an electron-donating –OMe group at the para-position, was used, a lower selectivity was observed, giving rise to a mixture of 2-quinolinones 14ei and 14ei′ (eq. 3).

Sulfur-atom-containing heterocycles constitute an important class of heterocycles because of their broad range of bioactivities and useful material properties. Encouraged by the success of our C–H cyclization approach to the synthesis of N-heterocycles such as indazoles,22) indoles,25) and 2-quinolinones,30,39) we became interested in whether this intramolecular C–H amination process could be extended to C–S bond formation to prepare sulfur-based heterocycles. Indeed, when we started the study, the literature contained no examples of transition-metal-catalyzed C–S bond formation via C–H functionalization. In this section, details of our investigation of this subject are addressed.
3.1. Benzothiazole Synthesis44–46)The benzothiazole framework represents a privileged structure that exhibits a plethora of biological activities.47) The classical approach to the construction of this class of compounds involves the oxidative cyclization of thiobenzanilides with various oxidants, such as potassium ferricyanide (Jacobson method) and bromine (Hugershoff method).48) However, low functional-group tolerance is a major drawback of these methods. The use of stoichiometric or excess amounts of toxic reagents, such as metals or bromine, may also be disadvantageous. The palladium- or copper-catalyzed cyclization of N-(2-halophenyl)thiobenzamides provides another route to benzothiazoles.49–52) It is necessary to prefunctionalize starting materials in these reactions, which significantly limits the utility and applicability of this approach. To test the feasibility of C–S bond formation via C–H functionalization, we chose the reaction of thiobenzanilide (15a), which would result in a new approach for the construction of a benzothiazole nucleus.
On the basis of systematic screenings of reaction parameters such as palladium catalysts, reoxidants, additives, and solvents, the optimal reaction conditions were determined to be 10 mol % PdCl2/50 mol % CuI/200 mol % Bu4NBr in DMSO/N-methylpyrrolidone (NMP) (1 : 1). The cyclization of 15a smoothly proceeded at 120°C under these conditions and produced benzothiazole 16a in 74% yield44) (Chart 9). Although the precise role of Bu4NBr is not clear at this stage, its addition is critical to achieve efficient conversion.

The substrate scope of the benzothiazole synthesis is considerably broad. Various substituted 2-arylbenzothiazoles 16b–q (Table 4) with both electron-donating and electron-withdrawing groups were obtained in high yields using the optimized reaction conditions. A particularly appealing feature of this system is its functional-group compatibility; the alkoxycarbonyl group (entry 11), the cyano group (entries 10, 15, and 16), and the halogen atoms, including iodine (entries 4–8 and 14), are well tolerated during the reaction. The reactions of substrates that possess a substituent at the 3-position (15c and 15g) regioselectively occurred at the less sterically hindered 6-position; the corresponding benzothiazoles (16c and 16g) were exclusively obtained, and no regioisomers (7-substituted benzothiazoles) were observed (entries 2 and 6).
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/T4.png%22>');%0A%09%09%09newWindow.document.close();%0A%09%09) | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Product | 16 (R or R′) | Yield (%) | Entry | Product | 16 (R or R′) | Yield (%) |
| 1 |  | 16b (6-OMe) | 65 | 9 |  | 16j | 83 |
| 2 | 16c (5-OMe) | 81 | 10 |  | 16k | 78 | |
| 3 | 16d (4-OMe) | 34 | 11 |  | 16l | 78 | |
| 4 | 16e (6-Cl) | 74 | 12 |  | 16m (4′-OMe) | 56 | |
| 5 | 16f (6-Br) | >99 | 13 | 16n (3′-OMe) | 61 | ||
| 6 | 16g (5-Br) | 53 | 14a) |  | 16o | 71 | |
| 7 | 16h (6-I) | 73 | 15a) |  | 16p | 71 | |
| 8a) | 16i (4-F) | 75 | 16 |  | 16q | 75 | |
a) 6 h.
Although the previously mentioned benzothiazole synthesis is highly efficient in terms of the substrate scope and the yields, it suffers one drawback: the process requires the use of a considerable excess of reoxidants (50 mol % CuI and 200 mol % Bu4NBr). Subsequent attempts to overcome this disadvantage revealed that molecular oxygen (O2), which is nontoxic, easy to handle, readily available, and relatively inexpensive, can be successfully used as a reoxidant.45) The key to the success of such reactions is the addition of CsF, although its precise role has yet to be determined. The reaction of 15a smoothly proceeded when the 10 mol % PdCl2/O2 catalyst system and 50 mol % CsF were used, affording 16a in 73% yield (Chart 10). The substrate scope of this method was found to be similar to that of the previous PdCl2/CuI/Bu4NBr system; this method thus represents a more applicable, greener approach to the construction of benzothiazole frameworks.

Moreover, water proved to be a suitable reaction medium in the benzothiazole synthesis (Chart 11). Notably, the reactions proceeded under considerably milder conditions when water was used compared with conditions used with our two previously reported systems (40°C vs. 100–120°C).46) For some substrates, the addition of an amphiphilic surfactant significantly enhanced the process. Indeed, the method represents a rare example of palladium-catalyzed C–H functionalization processes performed in water.53–55)

Multisubstituted benzo[b]thiophenes also constitute an important class of S-heterocycles, which exhibit various biological activities as well as properties useful in materials science.56) A number of methods for the synthesis of benzo[b]thiophenes have been reported in recent years, most of which involve the cyclization of benzenethiol derivatives. However, facile and versatile methods for accessing multisubstituted benzo[b]thiophenes are still limited. Furthermore, catalytic cyclization approaches involving transition metals for the construction of the benzo[b]thiophene skeleton, which would provide a more efficient and practical route, are extremely rare in the literature presumably because of catalyst poisoning by sulfur. Only a few reports in which gold57) or palladium58) catalysts have been used to effect C–S bond formation have appeared.
To establish a novel, catalytic synthetic procedure for benzo[b]thiophenes, ethenethiols 17 were chosen as starting materials for C–H cyclization. Systematic studies to determine the optimal reaction conditions surprisingly revealed that the palladium-catalyzed C–H cyclization of 1,2,2-triphenylethenethiol 17a more efficiently proceeded in the absence of reoxidants. The reaction of 17a using 25 mol % Pd(OAc)2 and 100 mol % Cu(OAc)2 as a catalyst system in DMSO at 80°C resulted in the formation of 18a in only 8% yield, whereas only using Pd(OAc)2 as the catalyst delivered 18a in 22% yield (Chart 12). In both cases, a small but not negligible amount of disulfide 19a was formed.

A subsequent examination of the reaction parameters revealed the optimized conditions, such as 10 mol % PdCl2 in DMSO at 120°C, which provided 18a in high yield (Table 5, entry 1). Several types of ethenethiols 17b–h (symmetrical substrates) were also used in this process by employing the established optimal conditions (entries 2–8). Yields were generally high, although the reaction of 17h, which possesses an ortho-methoxy group on each Ar1, was rather sluggish (entry 8).
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/T5.png%22>');%0A%09%09%09newWindow.document.close();%0A%09%09) | ||||
|---|---|---|---|---|
| Entry | Substrate | Product | 18 | Yield (%) |
| 1 | Ar1=Ar2=Ph (17a) |  | 18a | 82 |
| 2 | Ar1=Ph, Ar2=(4-OMe)C6H4 (17b) | 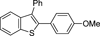 | 18b | 89 |
| 3 | Ar1=(4-Me)C6H4, Ar2=Ph (17c) |  | 18c | 77 |
| 4 | Ar1=(4-OMe)C6H4, Ar2=Ph (17d) |  | 18d | 74 |
| 5 | Ar1=(4-F)C6H4, Ar2=Ph (17e) |  | 18e | 72 |
| 6 | Ar1=(4-Cl)C6H4, Ar2=Ph (17f) |  | 18f | 85 |
| 7 | Ar1=(3-OMe)C6H4, Ar2=Ph (17g) |  | 18g | 72 |
| 8 | Ar1=(2-OMe)C6H4, Ar2=Ph (17h) |  | 18h | <16 |
The reaction of 17i, which has only one methoxy-substituted benzene ring (i.e., an unsymmetrical substrate), was also examined by employing our optimized reaction conditions. Despite the starting 17i being a single isomer, the cyclized product obtained was a mixture of two benzo[b]thiophenes, 18i-A and 18i-B, which suggests that the E/Z-isomerization of ethenethiol may occur relatively easily during the process (Chart 13).

Although extensive mechanistic studies have not yet been conducted, we believe that this palladium-catalyzed C–H cyclization proceeds via the formation of disulfide 19 (Chart 14). DMSO by itself 59–61) or in combination with various acidic co-reagents62–64) mediates the transformation of thiols to disulfides. The palladium catalyst may also be involved in this oxidation step, because the heating of 17a in DMSO at 120°C in the absence of palladium resulted in the formation of disulfide 19a in only 11% yield.62,63,65–67) Disulfide 19 formed from the corresponding ethenethiol 17 may undergo oxidative addition to palladium, leading to complex 20.68,69) Subsequent C–H cyclization can give rise to the desired benzo[b]thiophene 18.

The previously proposed mechanism complements the following observations: (1) the formation of a small amount of a disulfide compound was observed in most cases; (2) oxidants are not necessary for the process; and (3) isolated disulfide 19a can be converted into benzo[b]thiophene 18a in the presence of the palladium catalyst70) (Chart 15).

We investigated the synthesis of heterocyclic compounds by palladium-catalyzed C–H functionalization/intramolecular C–N (or C–S) formation. By this method, we efficiently obtained a series of N-heterocycles (indazoles, indoles, and 2-quinolinones) and S-heterocycles (benzothiazoles, benzo[b]thiophenes). Moreover, auto-tandem-type, one-pot synthesis of 2-quinolinones involving an oxidative Heck reaction followed by C–H cyclization using a palladium catalyst was also realized. Good-to-high yields, good functional-group tolerance, and wide substrate scopes were observed for each process, which demonstrates that our approach for the construction of heterocyclic frameworks is highly efficient and applicable. Indeed, the results shown in this review clearly indicate that the C–H cyclization strategy can provide novel, direct, and useful routes to biologically important heterocyclic frameworks. We believe that subsequent studies will further highlight the usefulness of this methodology in synthetic organic chemistry.
All the research in this review was conducted at the Graduate School of Pharmaceutical Sciences, Tohoku University. The author is very grateful to Emeritus Professor Takao Sakamoto, Professor Takayuki Doi, Professor Kou Hiroya, and Professor Yoshinori Kondo for their continuous support and helpful discussions. The author would also like to express his sincere appreciation to all of the students whose names are acknowledged in our publications cited in this review. This study was supported in part by a Grant-in-Aid for Young Scientists (B) from the Japan Society for the Promotion of Science (JSPS) and an Astellas Pharma Inc. Award in Synthetic Organic Chemistry, Japan.