Notes
Synthesis and Neuroprotective Effects of the Fluorine Substituted Salidroside Analogues in the PC12 Cell Model Exposed to Hypoglycemia and Serum Limitation
2013 年 61 巻 11 号 p. 1192-1196
詳細
2013 年 61 巻 11 号 p. 1192-1196
Salidroside (Sal) is a natural antioxidant extracted from the root of Rhodiola rosea L., a traditional Chinese medicinal plant, which elicits neuroprotective effects in the treatment of ischemic stroke. In an attempt to improve its neuroprotective effects, fluorine substituted Sal analogues were synthesized and their neuroprotective activities against the hypoglycemia and serum limitation induced cell injury in differentiated PC12 cells were evaluated. The target compounds displayed strong protective effects on the cell viability against the damage caused by hypoglycemia and serum limitation, especially for D1, which had a great potency superior to Sal and efficiently inhibited hypoglycemia and serum limitation induced cell nuclear morphologic changes and the increased apoptotic rate in a dose-dependent manner. These new findings may provide potentially important information for further development of Sal analogues and lay the basis for further studies on the cerebral ischemic stroke and neurodegenerative diseases for human clinical treatment.
Focal cerebral ischemia or stroke remains has been a major cause of death and disability in clinical practice, especially in the elderly population. Wide variety of cellular and molecular mechanisms involved in ischemic brain damage.1) Ischemia is characterized by lacking the blood flow to the brain, which results in insufficient supply of glucose, oxygen, serum and nutrient energy generation. Morphological manifestations of programmed cell death in the ischemic brain and biochemical evidence have revealed that apoptosis was implicated in ischemic stroke.2) Thus, the prevention and inhibition of neuronal apoptosis may be a reasonable choice for the treatment of permanent neurological damage for cerebral ischemia.
2-(4-Hydroxyphenyl)ethyl β-D-GlucopyranosideSalidroside (Sal), 2-(4-hydroxyphenyl)ethyl β-D-glucopyranoside (Fig. 1A), belonging to the family Crassulaceae, is one of the active ingredients extracted from the root of Rhodiola rosea L. R. rosea, mainly in the mountainous areas of a plant in high altitude areas in the Northeast Asia, has been used as traditional Tibetan medicine for a long time. The plant also shows a range of pharmacological properties, including antiaging, antiinflammation, antifatigue, anticancer, and cardioprotection.3–8) Recently, more attention has been focused on the study of its neuroprotective effect.9,10) However, the natural Sal in R. rosea production is low (<1.99%), and its neuroprotective effect for treatment cerebral ischemia was limited. Therefore, the development of novel Sal analogues with stronger neuroprotective effects by inhibiting neuronal apoptosis will be of great significance. Our previous studies have revealed that Sal analogues exhibited protective effects on hypoglycemia and serum limitation-induced cell apoptosis in cultured PC12 cells.11–13)
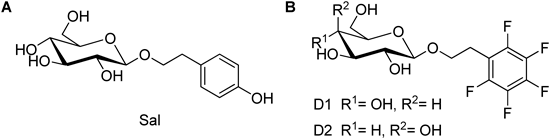
To develop new Sal analogues for human use, we were inspired by the fact that introducing fluorine atoms into drugs can alter the rate and route of drug metabolism14) and stereoelectronic factors associated with the fluorine atom can lead to changes in the biological action of molecules in comparison to its analogues substituted with hydroxy groups or hydrogen atoms.15) In addition, the small size of the fluorine atom, the enhanced lipophilicity it imparts to the molecules and the electronegativity of the atom often results in improved therapeutic drugs.16) Therefore, hydroxy group or hydrogen atoms of Sal’s aromatic ring were substituted with fluorine atoms so as to discover and develop of new neuroprotective effect agents for human use. The fluorine substituted Sal analogues (D1, D2, see Fig. 1B) were synthesized, and their neuroprotective activities and apoptosis effects in the cell injury induced by hypoglycemia and serum limitation were investigated and compared with Sal.
The synthetic route of Sal’s analogue (D1, D2) was outlined in Chart 1. The starting material 1-bromo-2,3,4,5,6-pentafluorobenzene (1) was converted to 2-(pentafluorophenyl)ethanol (2) by treatment with n-BuLi/hexane and ethylene oxide at −78°C in 76% yield. Compound 2 was treated with tetra-O-acetyl-α-D-glucopyranosyl bromide or tetra-O-acetyl-α-D-galactopyranosyl bromide in the presence of AgCO3 to give intermediate 3a or 3b, which were deacetylated with K2CO3 to yield the targeted compound D1 or D2. The structure of D1 and D2 were confirmed by of MS, IR, 1H-NMR spectra and elemental analysis (EA).

Reagents and conditions: (a) n-BuLi/hexane, ethylene oxide, ether, −78°C, 2 h; (b) tetra-O-acetyl-α-D-glucopyranosyl bromide or tetra-O-acetyl-α-D-galactopyranosyl bromide, Ag2CO3, CH2Cl2, molecular sieve powder, rt, overnight; (c) K2CO3, CH3OH, rt, 2 h.
The neuroprotective effects of target compounds D1 and D2 were investigated on preventing decreased cell viability in PC12 cells induced by hypoglycemia and serum limitation using Sal as positive controls. The results of these studies are summarized in Fig. 2. Hypoglycemia and serum limitation led to significant reduction of cell viability to 38.12±0.87% for 48 h, compared to control. When cells pretreated with D1, D2 and Sal under different concentration (20, 100, 300 µg/mL) for 24 h followed by hypoglycemia and serum limitation treatment, we found that D1 and D2 exhibited significant protective effects, which increased cell viability to 81.32±2.31% and 70.27±2.68%, respectively, which superior to Sal group (65.62±3.87%) at the same concentration of 300 µg/mL. Especially, compound D1 exhibited the most significant protective effect, which increased cell viability to 81.32±2.31%, 68.39±1.56% and 47.92±1.31% at the different concentration of 20, 100, 300 µg/mL, and were significantly higher than Sal and D1 groups respectively. One plausible explanation was that the glucose fragment of D1 may contribute to its bioactivities superior to D2 with galactose fragment. Furthermore, the protective activities of D1 and D2 with fluorine atoms substituted aromatic ring were also better than that of phenethyl analogues without phenolic hydroxyl which had been researched by our group.11) Therefore, these results suggested that the fluorine substituted Sal analogues displayed neuroprotective activities in the cell injury induced by hypoglycemia and serum limitation. D1 was further researched by Hoechst 33342 staining and flow cytometry with annexin V/PI staining.
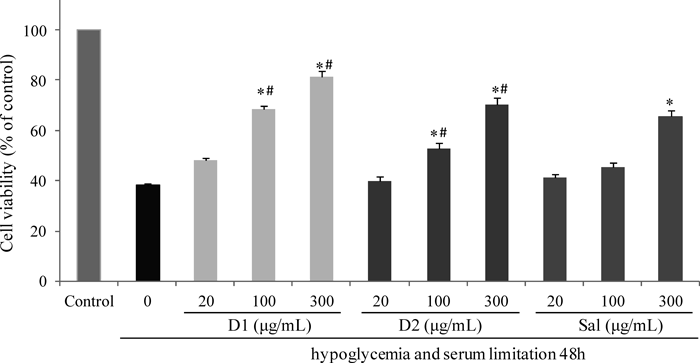
PC12 cells were pretreated with test compounds on different concentrations (20, 100, 300 µg/mL) for 24 h, then exposed to hypoglycemia and serum limitation for an additional 48 h with respective original concentrations of compounds maintained. Viability was calculated as the percentage of living cells in treated cultures compared to those in control cultures by using MTT assay. * p<0.05, versus exposure to hypoglycemia and serum limitation for 48 h in alone. # p<0.05, versus the positive control group Sal (300 µg/mL). The data were presented as mean±S.D. of three independent experiments (each in triplicate).
Apoptosis after cerebral ischemia is one of the major pathways that lead to the process of cell death.17) Therefore, morphological examinations using Hoechst 33342 staining were performed to determine the anti-apoptotic effects of the target compound D1. According to Fig. 3A, there was only 3.1±1.3% of the cells in the control group that underwent apoptosis or cell death, whereas hypoglycemia and serum limitation induced apoptotic/necrotic cells to 18.9±5.2%. However, treatment of Sal (100, 300 µg/mL) significantly decreased hypoglycemia and serum limitation induced apoptotic/necrotic cells to 10.8±1.9% and 8.2±2.0%; simultaneously treatment of D1 (100, 300 µg/mL) significantly decreased hypoglycemia and serum limitation induced apoptotic/necrotic cells to 8.4±2.1% and 3.8±1.9% (Fig. 3B). Interesting, treatment of D1 (300 µg/mL) significantly was better protective effect than Sal group with the same dose.
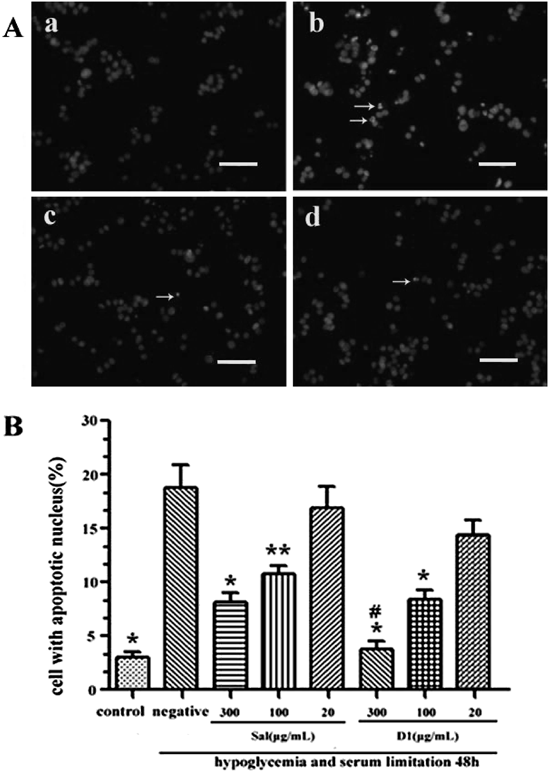
Scale bar for (a–d), 10 µm. (B) The number percentage of apoptotic cells in the total cell population is shown for six different treatments of PC12 cells. * p<0.01 and ** p<0.05 vs. hypoglycemia and serum limitation alone. # p<0.01 vs. Sal (300 µg/mL) group.
To further determine the anti-apoptotic effects of the target compound D1, flow cytometry with annexin V/PI staining provided further evidence that Sal pretreatment prevented hypoglycemia and serum limitation induced apoptosis in cultured PC12 cells. The percentages of apoptotic cells were determined by flow cytometry analysis in Fig. 4A. The quantitative comparison indicated that the percentages of early and late apoptotic rate in PC12 cells induced by exposure to hypoglycemia and serum limitation alone were 29.63% and 15.16% respectively. While PC12 cells were pretreated with 300 µg/mL of D1 prior to hypoglycemia and serum limitation, early stage and late stage of apoptosis were surprisingly prevented, and the apoptotic rate significantly decreased to 12.08% and 6.61%, which were better than that induced by pretreatment with Sal at the same concentration (Fig. 4B). The above results revealed that the target compound D1 could prevented the increased apoptotic rate and nuclear morphologic changes induced by hypoglycemia and serum limitation.

(A) PC12 cells in control group, hypoglycemia and serum limitation alone group, pretreatment with Sal (300 µg/mL) or D1 (300 µg/mL) plus exposure to hypoglycemia and serum limitation group were stained with FITC-Annexin V and PI, followed by flow cytometry analysis. (B) Quantitative analysis of the percentage of apoptotic and necrotic cells in total cell population were shown after different treatments. Data are expressed as means±S.D. of the percentages of apoptotic cells from three independent experiments. * p<0.01 vs. exposure to hypoglycemia and serum limitation alone. # p<0.01 vs. Sal (300 µg/mL) group.
In conclusion, two fluorine substituted Sal analogues D1 and D2 were synthesized, and their neuroprotective activities against the hypoglycemia and serum limitation-induced injury of differentiated PC12 cells were studied. Both D1 and D2 displayed strong protective effects on the cell viability against the damage caused by hypoglycemia and serum limitation, especially for D1, which had a great potency superior to Sal and efficiently prevented the increased apoptotic rate and nuclear morphologic changes induced by hypoglycemia and serum limitation. Although a detailed molecular mechanism that links all these events should be further investigated, the present new findings may provide potentially important information for further development of Sal analogues and lay the basis for further studies on the cerebral ischemic stroke and neurodegenerative diseases for human clinical treatment.
Melting points were determined on a Mel-TEMP II melting point apparatus and uncorrected. Optical rotations were measured with a JASCO P-1020 polarimeter (cell length: 100 mm). Infrared (IR) spectra (KBr) were recorded on a Nicolet Impact 410 instrument (KBr pellet). 1H-NMR spectra were recorded with a Bruker Avance 300 MHz spectrometer at 300 K, using tetramethylsilane (TMS) as an internal standard. MS spectra were recorded on a Shimadzu GC-MS 2010 (electron ionization (EI)) or a Mariner Mass Spectrum (electrospray ionization (ESI)). Element analysis was performed on an Eager 300 instrument. All compounds were routinely checked by TLC and 1H-NMR. TLCs and preparative thin-layer chromatography were performed on silica gel GF/UV 254, and the chromatograms were conducted on silica gel (200–300 mesh) and visualized under UV light at 254 and 365 nm. All solvents were reagent grade and, when necessary, were purified and dried by standards methods. Solutions after reactions and extractions were concentrated using a rotary evaporator operating at a reduced pressure of ca. 20 Torr. Organic solutions were dried over anhydrous sodium sulfate. Compound 1 was commercially available, and tetra-acetyl glucosyl bromide and tetra-acetyl galactosyl bromide were prepared according to the literature.18,19)
Synthesis of D1 and D22-(Pentafluorophenyl)ethanol (2): 1-Bromo-2,3,4,5,6-pentafluorobenzene (1, 23.0 g, 93 mmol) was dissolved in ether (30 mL) and cooled to −78°C. To this solution, n-BuLi (1.6 mol/L solution in hexanes, 60 mL, 96 mmol) was slowly added. After 1 h, ethylene oxide in toluene solution was added to the reaction mixture, and the flask was removed from the cooling bath and the mixture was stirred at rt for 1 h. After saturated aqueous NH4Cl solution (100 mL) was added, the product was extracted with diethyl ether (2×300 mL). The combined organic layers were dried over magnesium sulfate and evaporated in vacuo. The resultant yellow residue was purified by rapid column chromatography (AcOEt–PE=1 : 10–1 : 5, v/v) to afford colorless oil (15.0 g, yield: 75%).
2-(Pentafluorophenyl)ethyl 2,3,4,6-Tetra-O-acetyl-β-D-glucopyranoside (3a): To CH2Cl2 (10 mL) solution of 2 (0.76 g, 3.6 mmol) were added Ag2CO3 (2.00 g, 7.2 mmol) and molecular sieve powder (1.2 g) by nitrogen protection, and the mixture was stirred away from light for 0.5 h at room temperature, then tetra-O-acetyl-α-D-glucopyranosyl bromide (2.95 g, 7.2 mmol) was added quickly, and continuously stirred overnight. The precipitate was filtrated and the filtrate was concentrated in vacuo, and the residue was purified by rapid column chromatography (PE–EtOAc=1 : 2–1 : 3, v/v) to afford 3a as colorless viscous liquid; yield: 75%.
2-(Pentafluorophenyl)ethyl 2,3,4,6-Tetra-O-acetyl-β-D-galactopyranoside (3b): Compound 3b was synthesized from 2 (0.76 g, 3.6 mmol) and tetra-O-acetyl-α-D-galactopyranosyl bromide (2.95 g, 7.2 mmol) according to the synthetic procedure of 3a in yield 72%, colorless viscous liquid.
2-(2,3,4,5,6-Pentafluorophenyl)ethyl β-D-Glucopyranoside (D1): To 3a (1.14 g, 2.1 mmol) in MeOH (20 mL) was added K2CO3 (0.31 g, 2.3 mmol) to pH=8–10 and the mixture was allowed to stir for 2 h at room temperature. The solution was neutralized with Amberlite IRA-120 (H+) resin to pH 7, then filtrated and concentrated in vacuo, and the residue was purified by rapid column chromatography (MeOH–CHCl3=1 : 10–1 : 5, v/v) to afford white solid D1; yield: 85%; mp: 134–136°C; [α]D26.9 −18.5 (c=0.15, MeOH). IR (KBr, ν, cm−1): 3406, 2940, 1507, 1120, 1094, 1037. 1H-NMR (CD3OD, 300 MHz): δ (ppm) 3.06 (t, J=6.9 Hz, 2H), 3.13 (t, J=8.4 Hz, 1H), 3.23–3.29 (m, 3H), 3.63 (t, J=5.7 Hz, 1H), 3.74–3.86 (m, 2H), 4.06 (t, J=6.9 Hz, 1H), 4.45 (d, J=8.4 Hz, 1H). MS (ESI) m/z=375 [M+H]+; Anal. Calcd for C14H15F5O6: C, 44.93; H, 4.04. Found: C, 44.99; H, 4.06.
2-(2,3,4,5,6-Pentafluorophenyl)ethyl β-D-Galactopyranoside (D2): Compound D2 was synthesized from 3b (1.14 g, 2.1 mmol) and K2CO3 (0.31 g, 2.3 mmol) according to the synthetic procedure of D1 in yield 78%, white solid. mp 110–113°C; [α]D26.9 −8.2 (c=0.11, MeOH). IR (KBr, ν, cm−1): 3385, 2935, 1506, 1121, 1085. 1H-NMR (CD3OD, 300 MHz): δ (ppm) 3.06 (t, J=7.0 Hz, 2H), 3.43–3.50 (m, 3H), 3.65–3.70 (m, 2H), 3.73–3.81 (m, 2H), 4.01–4.09 (t, J=6.9 Hz, 1H), 4.40 (d, J=8.4 Hz, 1H). MS (ESI) m/z=375 [M+H]+; Anal. Calcd for C14H15F5O6: C, 44.93; H, 4.04. Found: C, 44.89; H, 4.04.
Cell TreatmentPC-12 cells, obtained from the American Type Culture Collection (Manassas, VA, U.S.A.), were plated and maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum, 10% horse serum, 100 U/mL streptomycin, and 100 U/mL penicillin in a humidified atmosphere of 5% CO2 and 95% air at 37°C. The cells were differentiated by treating with nerve growth factor (NGF) (50 ng/mL) every other day for 6 d.
PC12 cells were pre-incubated with D1, D2 or Sal for 24 h, then were exposed to hypoglycemia and serum limitation, with the glucose-free DMEM supplemented including 1% fetal bovine serum, 1% horse serum, 100 U/mL streptomycin, and 100 U/mL penicillin, for an additional 48 h with original concentrations. At the same time, treatment only with culture medium containing high glucose/enough serum or only exposure to hypoglycemia and serum limitation (but without pretreatment) was considered control group or negative group. At the end of cell treatments, we carried out the following different tests.
Cell Viability Assay3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was used to detect cell viability as described previously. MTT was dissolved in DMEM medium and added the cell samples to make final concentration of 0.5 mg/mL at 37°C for 4 h. Afterward, medium was added to a solution of 20% sodium dodecyl sulfide (SDS) to dissolve formazan generated. The absorbance (optical density (OD)) values were measured by spectrophotometry at 570 nm with an EIX-800 Microelisa reader (Bio-Tek Inc., U.S.A.).
Hoechst 33342 Staining AssaysThe PC12 cells were fixed in 4.0% paraformaldehyde for 20 min, and then stained with 5 µg/mL Hoechst 33342 dye at 37°C for 10 min, followed by observation under a DMR fluorescence microscope (Leica Microsystems, Wetzlar, Germany) with ultraviolet illumination. The dye was excited at 340 nm, and emission was filtered with a 510 nm barrier filter. In order to quantity the apoptotic process, cells with fragmented or condensed DNA and normal DNA were counted, respectively. The data were expressed as a percent of control value and mean±S.E.M. of three experiments.
Flow Cytometry with Annexin V/PI StainingApoptotic/necrotic cells were measured by flow cytometry as described previously.20) The PC12 cells were harvested, was hed cells twice with cold PBS and then resuspend cells in 1X Binding Buffer at a concentration of 1×106 cells/mL per assay. Transfer 100 µL of the solution (1×105 cells) to a 5 mL culture tube, 5 µL of fluorescein isothiocyanate (FITC)-conjugated Annexin V and 5 µL propidium iodide (PI) were added, then Gently vortexed the cells and incubated for 15 min at RT (25°C) in the dark, and added 400 µL of 1X Binding Buffer to each tube. Cells were analyzed by flow cytometry within 1 h (BD, CA).
Statistical AnalysisAll Data were expressed as means±S.E.M. Statistical differences between groups were analyzed by oneway analysis of variance with subsequent Turkey’s tests. In all cases, p<0.05 was considered statistically significant.
We gratefully acknowledge the financial support by the Natural Science Foundation of China (Grant Nos. 21242005 and 81171457), Biotechnology and New Medicine Projects of Nantong City (AS2011016), Nature Science Foundation of Nantong University (Grant Nos. 10Z074 and 12ZY035), and also thank a project funded by the Priority Academic Programs Development of Jiangsu Higher Education Institutions (PAPD).