Regular Articles
Synthesis of Am80 (Tamibarotene) Prodrug Candidates, Congeners and Metabolites
2013 Volume 61 Issue 8 Pages 846-852
Details
2013 Volume 61 Issue 8 Pages 846-852
Compound 1 (IT-M-07000) was previously reported as a candidate prodrug of Am80 (Tamibarotene; used to treat acute promyelocytic leukemia), and shown to be efficiently metabolized to Am80 via β-oxidation. Here, we describe in detail the synthesis of 1, together with another tetradeuterated candidate prodrug, IT-YA-00616 (2), as well as two congeners, and several metabolic intermediates of 1 previously detected in mouse plasma.
Am80 (Tamibarotene) is a synthetic retinoic acid receptor (RAR)-α,β-selective agonist that does not bind to the proinflammatory RAR-γ subtype or to retinoid X receptors (RXRs). Because of this activity profile, Am80 does not induce unfavorable side-effects caused by RAR pan-agonists, such as all-trans retinoic acid (ATRA)1) (Fig. 1). It is also characterized by high stability to light, heat, and oxidation in air, unlike retinoic acid. Am80 was approved in Japan in 2005 as a therapeutic agent for recurrent refractory acute promyelocytic leukemia (APL).2,3) Furthermore, it has recently been suggested that stimulation of the RARα and RARβ signaling pathway with agonists enhances clearance of amyloid β, so that retinoids, including Am80, may be promising candidates for treatment of Alzheimer’s disease.4,5) However, in the required case of long-term administration of retinoids such as Am80, the patients are consistently threatened by the side-effects endemic to retinoids (retinoic acid syndrome): dyspnea, fever, weight gain, hypotension, and pulmonary infiltrates.6)

With these facts in mind, we previously designed a candidate prodrug of Am80, aiming at sustained-release character and consequential reduction of the above described retinoic acid syndrome. We envisaged that IT-M-07000 (1) (Fig. 1), a two-carbon-elongated propionic acid derivative of Am80, would be readily metabolized to Am80 via the carboxylic acid β-oxidation pathway, which involves i) dehydrogenation by flavin adenine dinucleotide (FAD) to form α,β-unsaturated acyl-CoA, ii) hydration on the β-carbon to afford 3-hydroxyacyl-CoA, iii) oxidation by oxidized form of nicotinamide adenine dinucleotide (NAD+) to yield 3-ketoacyl-CoA, iv) thiolysis of 3-ketoacyl CoA by another molecule of coenzyme A.7,8) Sodium phenylbutyrate (Buphenyl®) is a typical example of a prodrug that utilizes this β-oxidation process to release sodium phenylacetate, which is clinically used for the treatment of urea cycle disorders.9) In our earlier work, we synthesized 1, and examined its disposition in mice, reporting its efficient metabolism to Am80, and identifying the metabolic intermediates of the β-oxidation pathway.10) We further showed that administration of 1 to mice resulted in larger area under the curve (AUC), lower Cmax, and longer t1/2, mean residence time (MRT) of Am80 derived from 1, compared with those after administration of Am80 itself. These results confirmed that 1 is a promising candidate for an Am80 prodrug with reduced side effects and longer duration of drug efficacy.
In the present paper, we describe in detail the synthesis of 1 and its metabolic intermediates. We also synthesized IT-YA-00616 (2) (Fig. 1), the 2,2,3,3-tetradeuterated derivative of 1, as another candidate prodrug of Am80, aiming at further improvement of the pharmacokinetic parameters, as well two congeners, the 2,2- and 3,3-dideuterated compounds, for examination of the isotope effect.
Our first Am80 prodrug candidate 1 was readily prepared via two routes, A and B (Chart 1).

(a) SOCl2, benzene, reflux; (b) 6-amino-1,2,3,4-tetrahydro-1,2,3,4-tetramethylnaphthalene (4), Et3N, CH2Cl2, rt, 97% from 4; (c) LiOH, MeOH–DME–H2O (3 : 2 : 1), reflux, 93% from 5, 98% from 9; (d) ClCOOMe, Et3N, THF, −20°C, then NaBH4, H2O, 0°C–rt, 94%; (e) MnO2, CH2Cl2, reflux, 96%; (f) triethylphosphonoacetate, K2CO3, EtOH, 62–64°C, 97%; (g) H2, Pd/C, EtOH, rt, 99%.
Route A: Methyl 3-(4-carboxy)phenylpropionate (3)11,12) was treated with thionyl chloride, then condensed with 1,1,4,4-tetramethyl-1,2,3,4-tetrahydro-6-aminonaphthalene (4)13) in the presence of triethylamine to provide 5, which was easily hydrolyzed to 1. But, although this route is straightforward, compound 3 is not readily available.
Route B: Am80, which is readily available at this laboratory, was converted to α,β-unsaturated ester 8 by way of alcohol 6 and aldehyde 7 in three steps in a high overall yield. Hydrogenation of 8 followed by hydrolysis of the resulting 9 conveniently afforded 1.
Preparation of Metabolic Intermediates of 1We next planned to synthesize putative intermediates for confirmation of the metabolic disposition of 1. Chart 2 shows the expected metabolic pathway of 1 to Am80 via β-oxidation. Compounds 11′, 12′, and 13′, corresponding to hydrolyzed products of the CoA intermediates 11–13, respectively, were selected for synthesis. The α,β-unsaturated carboxylic acid 11′ was prepared by simple hydrolysis of compound 8. The anion of tert-butyl acetate was allowed to react with Am80 methyl ester 15 to yield β-ketoester 16 and by-product 17 (Chart 2). Treatment of 16 with trifluoroacetic acid (TFA) readily provided 13′. The remaining compound 12′ was acquired by reduction of 16 with sodium borohydride followed by TFA treatment of the resulting tert-butyl 3-hydroxypropionate 18.

(a) LiCH2COOt-Bu, THF, −78°C, 16 59%, 17 35%; (b) TFA, CH2Cl2, 0°C–rt, 13′ 84%, 12′ 91%; (c) NaBH4, EtOH, 0°C, 97%.
Among the three putative metabolic intermediates, 11′ and 12′ were confirmed to be identical with metabolites in plasma of mice treated with 1 by comparison of their retention times in HPLC and their MS spectra.10) Two other peaks were identified as 1 itself and Am80. However, 13′ was not detected in plasma, whole blood, or liver. After intravenous administration of 13′, only Am80, but not 13′ itself, was detected. These results suggest that 13′ is rapidly converted to Am80 in mouse.
Synthesis of IT-YA-00616 (2) and Its 3,3-Dideuterated Congener 30Based on the previous finding that 1 is a promising candidate as a prodrug of Am80, we considered a further modification in order to improve the pharmacokinetics. We hypothesized that deuteration of the ethylene moiety of 1, affording 2, would be effective, because the rate of a reaction involving a carbon–deuterium (C–D) bond is typically 2 to 10 times slower than that of the corresponding carbon–hydrogen (C–H) bond.14) Accordingly, introduction of deuterium was expected to improve the sustained-release characteristics, due to the kinetic isotope effect.
Initially, we developed a bulk preparation method of 3-(4-carboxy)phenylpropionic acid (21) (Chart 3) as the substrate for ethylene deuteration, because although 21 is commercially available, it is very expensive. Terephthalaldehydic acid 19 (0.8 M scale) was condensed with malonic acid in the presence of a catalytic amount of piperidine in hot pyridine to afford p-carboxycinnamic acid 20 in 98% yield.15) The desired 21 was readily obtained in 95% yield by hydrogenation (H2 0.3 MPa) of 20 over a palladium-carbon catalyst in the presence of sodium carbonate (1.15 eq) in water.
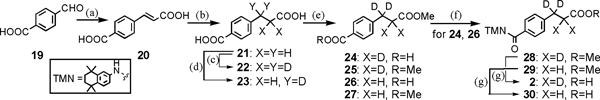
(a) Malonic acid, piperidine, pyridine, 80°C, 98%; (b) H2 (0.3 MPa), Pd/C, Na2CO3, H2O, 95%; (c) H2, D2O, Pd/C, Na2CO3, 130°C, 97%; (d) H2, D2O, Pd/C, Na2CO3, 100°C, 97%; (e) MeOH, cat. SOCl2 (1–2 mol%), rt, 24 88%, 25 4%, recovery 1% from 22 (0.10 mol) and 26 86%, 27 6%, recovery 1% from 23 (14.5 mmol); (f) SOCl2, cat. DMF, benzene, reflux, then 3, Et3N, CH2Cl2, rt, 28 96% from 24, 29 97% from 26; (g) LiOH, MeOH–DME–H2O (3 : 2 : 1), reflux, 2 98%, 30 98%.
Our next task was deuteration of 21 employing the Pd/C–H2–D2O system reported by Sajiki and colleagues.16) (Chart 3). After optimization of temperature, reaction time, amount of palladium catalyst, initial hydrogen pressure, and reaction vessel, we eventually obtained the desired 22 in high yield (29.8 g, 97% yield, D ratio of X=97–98%, Y=98%) from 21 (30.0 g, 0.155 mol) in a globular shape reaction vessel at 130°C (see Experimental). It is noteworthy that only partial deuteration occurred to provide the 3,3-d2 product 23 (D ratio of X=0, Y>98%) when the reaction was carried out in a Parr reaction vessel (circular cylinder) at 100°C. We found that the shape of the reaction vessel strongly influenced the deuteration ratio even under the other conditions such as temperature, reaction time were identical.
With the requisite deuterated compound 22 in hand, the prodrug candidate 2 and the corresponding 3,3-dideuterated 30 were synthesized (Chart 3). Monoesterification of the dicarboxylic acids 22 and 23 was carried out with a catalytic amount of thionyl chloride in methanol.12) By-product diester (25, 27, respectively) and a mixture of acidic compounds (24 and recovered 22, 26 and recovered 23, respectively) were extractively separated and the starting material 22 or 23 was conveniently recovered from the mixture by simple filtration of a slightly slurried dichloromethane solution. The desired 24 and 26 were easily isolated in excellent yields by evaporation of the respective filtrates and recrystallization of the residues from dichloromethane–hexane. These monocarboxylic acids were readily led to 2 and 30 by way of esters 28 and 29, respectively, according to the method already shown in Chart 1.
Preparation of 2,2-Dideuterated Congener 32The unexpectedly acquired compound 30 was expected to be useful for pharmacokinetic profiling of 2. Therefore, we also synthesized the other 2,2-dideuterated congener 32, by reductively condensing aldehyde 7 with Meldrum’s acid according to Ramachary’s method17) to give 31, which was readily transformed to 32 in refluxing dry pyridine in the presence of D2O18) (Chart 4).

(a) Meldrum’s acid, Hantzsch ester, L-proline, CH3CN, rt, 85%; (b) D2O, pyridine, reflux, 80%.
We present full details of the synthesis of two Am80 prodrug candidates, IT-M-07000 (1) and IT-YA-00616 (2). Compound 1 was previously characterized as a promising Am80 prodrug. The new candidate 2, corresponding to tetradeuterated 1, is expected to show a superior pharmacokinetic profile to 1 as an Am80 prodrug, owing to the deuterium isotope effect. We also synthesized two other congeners (30, 32), and several metabolic intermediates of 1 previously detected in mouse plasma. Mechanistic and kinetic studies of 2 are in progress.
Melting points were determined on a Yanagimoto micro-melting point apparatus (hot plate), and are not corrected. MS and high-resolution MS (HR-MS) were recorded on a Hitachi M-80B spectrometer in a direct inlet (DI) mode at an ionizing voltage of 70 eV, and figures in parentheses indicate the relative intensities. IR spectra were measured on a Shimadzu IR-460 spectrophotometer. 1H-NMR spectra were obtained on a Varian Mercury 300 (300 MHz) and coupling constants (J values) are rounded to the nearest 0.5 Hz. 13C-NMR spectra were measured on a Varian Mercury 300 (75 MHz) under proton-decoupled conditions. Column chromatography was conducted on silica gel (SiO2, Fuji Davison BW 200), and preparative TLC (PTLC) was carried out on glass plates (20×20 cm) coated with Merck Silica gel 60PF254 (0.8 mm thick) unless otherwise specified, and the developing solvent is indicated in parentheses. Usual work-up refers to washing of the organic layers with water or brine, drying over anhydrous Na2SO4, and evaporating off the solvents under reduced pressure. Tetrahydrofuran (THF) was distilled from sodium/benzophenone ketyl prior to use.
Synthesis of 1Methyl 3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propionate (5): SOCl2 (0.65 mL, 8.91 mmol) was added to a slurry of 3 (460 mg, 2.21 mmol) in benzene (6 mL), and the mixture was refluxed with stirring for 1.5 h. The volatile materials were evaporated off, then benzene (4 mL) was added to the resulting residue and evaporated again to dryness. The residue was dissolved in CH2Cl2 (2 mL) and the solution was cooled in an ice bath. A solution of 4 (359 mg, 1.77 mmol), Et3N (0.98 mL, 7.04 mmol) in CH2Cl2 (4 mL) was added to this and the mixture was stirred at an ambient temperature for 18 h. Saturated NaHCO3–H2O was added and the whole was extracted with EtOAc. Usual work-up and purification by column chromatography (CHCl3) gave 5 (674 mg, 97% from 4) as colorless prisms, mp 135–136°C (CH2Cl2–hexane). HR-MS Calcd for C25H31NO3: 393.2302. Found: 393.2315. MS (m/z): 393 (M+, 35), 378 (100), 362 (5), 191 (76), 131 (35), 103 (18), 91 (10), 59 (7), 43 (10). IR (KBr) cm−1: 1707, 1659. 1H-NMR (CDCl3) δ: 1.28 (6H, s), 1.30 (6H, s), 1.69 (4H, s), 2.67 (2H, t, J=7.5 Hz), 3.02 (2H, t, J=7.5 Hz), 3.68 (3H, s), 7.30 (1H, d, J=8.5 Hz), 7.32 (2H, A2B2, J=8 Hz), 7.41 (1H, dd, J=8.5, 2.5 Hz), 7.53 (1H, d, J=2.5 Hz), 7.69 (1H, br s, CONH), 7.80 (2H, A2B2, J=8 Hz).
3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propionic Acid (IT-M-07000, 1): The compound 5 (525 mg, 1.34 mmol) was dissolved in MeOH–1,2-dimethoxyethane (DME)–H2O (3 : 2 : 1, 9 mL) and LiOH·H2O (85 mg, 2.02 mmol) was added and the mixture was stirred under reflux for 2.5 h. The reaction was quenched by addition of HCl–H2O (1 N, 2.10 mL, 2.10 mmol) and the whole was extracted with EtOAc. Usual work-up followed by recrystallization afforded 1 (470 mg, 93%) as colorless fine needles, mp 230–231°C (EtOAc). Anal. Calcd for C24H29NO3: C, 75.96; H, 7.70; N, 3.69. Found: C, 76.17; H, 7.63; N, 3.76. HR-MS Calcd for C24H29NO3: 379.2146. Found: 379.2131. MS (m/z): 379 (M+, 33), 364 (100), 177 (92), 131 (14), 107 (22), 103 (20), 77 (16). IR (KBr) cm−1: 1711, 1616. 1H-NMR (CDCl3) δ: 1.28 (6H, s), 1.30 (6H, s), 1.69 (4H, s), 2.72 (2H, t, J=7.5 Hz), 3.04 (2H, t, J=7.5 Hz), 7.30 (1H, d, J=8.5 Hz), 7.33 (2H, A2B2, J=8.5 Hz), 7.41 (1H, dd, J=8.5, 2 Hz), 7.53 (1H, d, J=2 Hz), 7.73 (1H, br s, CONH), 7.80 (2H, A2B2, J=8.5 Hz). 13C-NMR (DMSO-d6) δ: 30.2, 31.6, 31.7, 33.5, 34.0, 34.59, 34.63, 34.9, 118.0, 118.2, 126.4, 127.6, 128.2, 132.8, 136.7, 139.7, 144.5, 144.6, 165.1, 173.6.
4-Hydroxymethyl-N-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)benzamide (6): To a cooled (−20°C) solution of Am80 (320 mg, 0.91 mmol) in THF (5 mL), Et3N (152 µL, 1.09 mmol) was added a solution of ClCOOMe (82 µL, 1.06 mmol) in THF (2 mL) and the mixture was stirred at that temperature for 1 h. The mixture was filtered through a Celite bed and the Celite was rinsed with THF (3 mL). The filtrate and the THF wash were combined and the whole was cooled in an ice bath. After addition of NaBH4 (208 mg, 5.47 mmol), H2O (4 mL) was gradually added dropwise during 15 min with vigorous stirring, and the resulting mixture was further stirred at 0°C to an ambient temperature for 18 h. The reaction was quenched by addition of satd. NH4Cl–H2O and the whole was extracted with EtOAc. Usual work-up followed by column chromatography [hexane–EtOAc (2 : 1)] provided 6 (290 mg, 94%) as colorless prisms, mp 161–162.5°C (CH2Cl2–hexane). HR-MS Calcd for C22H27NO2: 337.2040. Found: 337.2045. MS (m/z): 337 (M+, 27), 322 (87), 135 (100), 107 (18), 89 (34), 77 (28). IR (KBr) cm−1: 1634. 1H-NMR (CDCl3) δ: 1.28 (6H, s), 1.30 (6H, s), 1.69 (4H, s), 1.93 (1H, t, J=4.5 Hz, OH), 4.78 (2H, d, J=4.5 Hz), 7.30 (1H, d, J=8.5 Hz), 7.43 (1H, dd, J=8.5, 2.5 Hz), 7.47 (2H, A2B2, J=8 Hz), 7.54 (1H, d, J=2.5 Hz), 7.76 (1H, br s, NH), 7.85 (2H, A2B2, J=8 Hz).
4-Formyl-N-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)benzamide (7): A slurry of 6 (1.891 g, 5.61 mmol) and MnO2 (3.905 g, 44.9 mmol) in CH2Cl2 (35 mL) was refluxed with stirring for 2 h. The mixture was filtered through a Celite bed and the Celite was washed with CHCl3. After evaporation of the solvent, resulting crystalline mixture was recrystallized to give 7 (1.802 g, 96%) as colorless prisms, mp 185.5–186.5°C (CH2Cl2–hexane). HR-MS Calcd for C22H25NO2: 335.1884. Found: 335.1913. MS (m/z): 335 (M+, 32), 320 (100), 133 (68), 105 (28), 77 (24), 51 (9). IR (KBr) cm−1: 1690, 1662. 1H-NMR (CDCl3) δ: 1.29 (6H, s), 1.30 (6H, s), 1.70 (4H, s), 7.33 (1H, d, J=8.5 Hz), 7.44 (1H, dd, J=8.5, 2 Hz), 7.53 (1H, d, J=2 Hz), 7.79 (1H, br s, NH), 7.99 (2H, A2B2, J=8.5 Hz), 8.03 (2H, A2B2, J=8.5 Hz), 10.10 (1H, s).
Ethyl (2E)-3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propenoate (8): K2CO3 (910 mg, 6.59 mmol) was added to a solution of 7 (1.765 g, 5.27 mmol) and triethyl phosphonoacetate (1.523 g, 6.80 mmol) in EtOH (40 mL), and the mixture was heated at 62–64°C for 3 h. After cooling, satd. NH4Cl–H2O was added and the whole was extracted with EtOAc. Usual work-up and column chromatography [hexane–EtOAc (39 : 1 to 14 : 1)] afforded 8 (2.076 g, 97%) as colorless needles, 166–166.5°C (CH2Cl2–hexane). HR-MS Calcd for C26H31NO3: 405.2302. Found: 405.2292. MS (m/z): 405 (M+, 31), 390 (100), 203 (60), 175 (22), 102 (29), 91 (20). IR (KBr) cm−1: 1697, 1663, 1644. 1H-NMR (CDCl3) δ: 1.28 (6H, s), 1.30 (6H, s), 1.35 (3H, t, J=7 Hz), 1.70 (4H, s), 4.29 (2H, q, J=7 Hz), 6.52 (1H, d, J=16 Hz), 7.31 (1H, d, J=8.5 Hz), 7.43 (1H, dd, J=8.5, 2.5 Hz), 7.53 (1H, d, J=2.5 Hz), 7.63 (2H, A2B2, J=8 Hz), 7.71 (1H, d, J=16 Hz), 7.74 (1H, br s, NH), 7.89 (2H, A2B2, J=8 Hz).
Ethyl 3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propionate (9): A slurry of 8 (1.010 g, 2.49 mmol) and Pd/C (10%, 25 mg, 23.5 mgatom) in EtOH (25 mL) was hydrogenated under H2 atmosphere (1 atm) at an ambient temperature for 2 h. Filtration through Celite bed, washing with CHCl3, and evaporation of the solvents left a crystalline residue which was recrystallized to provide 9 (1.007 g, 99%) as colorless fine needles, mp 132–132.5°C (CH2Cl2–hexane). HR-MS Calcd for C26H33NO3: 407.2459. Found: 407.2445. MS (m/z): 407 (M+, 36), 329 (100), 362 (5), 205 (61), 177 (12), 131 (37), 103 (18), 77 (10). IR (KBr) cm−1: 1724, 1642. 1H-NMR (CDCl3) δ: 1.24 (3H, t, J=7 Hz), 1.28 (6H, s), 1.30 (6H, s), 1.62 (4H, br s), 2.65 (2H, t, J=7.5 Hz), 3.02 (2H, t, J=7.5 Hz), 4.13 (2H, q, J=7 Hz), 7.30 (1H, d, J=8.5 Hz), 7.32 (2H, A2B2, J=8 Hz), 7.41 (1H, dd, J=8.5, 2.5 Hz), 7.53 (1H, d, J=2.5 Hz), 7.72 (1H, br s, NH), 7.79 (2H, A2B2, J=8 Hz).
Preparation of 1 from 9: In the completely same manner as for the preparation of 1 from 5, 9 (556 mg, 1.37 mmol) was hydrolyzed to give 1 (496 mg, 96%) as colorless needles.
Preparation of Metabolites(2E)-3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propenoic Acid (11′): In the same manner as for the preparation of 1 from 5, 8 (109 mg, 0.269 mmol) was hydrolyzed with LiOH·H2O (15 mg, 0.357 mmol) to leave a residue which was recrystallized to afford 11′ (96 mg, 95%) as colorless prisms, mp 226–227°C (CH2Cl2). HR-MS Calcd for C24H27NO3: 377.1989. Found: 377.1988. MS (m/z): 377 (M+, 29), 362 (100), 175 (96), 147 (18), 102 (17), 91 (41), 44 (32). IR (KBr) cm−1: 1682, 1646, 1625. 1H-NMR (CDCl3) δ: 1.29 (6H, s), 1.31 (6H, s), 1.70 (4H, s), 6.54 (1H, d, J=16 Hz), 7.32 (1H, d, J=8.5 Hz), 7.43 (1H, dd, J=8.5, 2.5 Hz), 7.53 (1H, d, J=2.5 Hz), 7.66 (2H, A2B2, J=8 Hz), 7.74 (1H, br s, NH), 7.81 (1H, d, J=16 Hz), 7.91 (2H, A2B2, J=8 Hz).
tert-Butyl 3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]-3-oxopropionate (16) and Di-tert-butyl 3-Hydroxy-3-[4-[(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]glutarate (17): n-BuLi (1.64 M/hexane, 2.00 mL, 3.28 mmol) was added to a cooled (−20°C) solution of i-Pr2NH (0.46 mL, 3.29 mmol) in THF (10 mL) and the mixture was stirred for 15 min. It was then cooled to −78°C and to this was added dropwise CH3COOtert-Bu (0.44 mL, 3.28 mmol) and stirred at the same temperature for 30 min. A solution of 15 (240 mg, 0.657 mmol) in THF (5 mL) was added to this and the whole was stirred at −78°C to an ambient temperature for 4 h. Quenching with satd. NH4Cl–H2O, extraction with EtOAc, usual work-up and purification by PTLC [hexane–EtOAc (10 : 1)] afforded 16 (175 mg, 59%) and 17 (129 mg, 35%) in order of increasing polarity. 16: Colorless foam. HR-MS Calcd for C28H35NO4: 449.2564. Found: 449.2568. MS (m/z): 449 (M+, 27), 434 (36), 360 (38), 334 (48), 173 (24), 147 (59), 104 (25), 59 (100), 57 (71), 43 (27), 41 (49). IR (CHCl3) cm−1: 1725, 1676. 1H-NMR of keto- and enol-form (ca. 2.7 : 1, CDCl3) δ: 1.20 (6H, s), 1.21 (6H, s), 1.35 and 1.47 (9H, s each), 1.61 (4H, br s), 3.85 (2H of keto form, s, D2O exchangeable), 5.57 (1H of enol form, s), 7.22 and 7.21 (1H, d each, J=8.5 Hz), 7.34–7.41 (1H, m), 7.45–7.51 (1H, m), 7.86 and 7.74 (2H, A2B2 each, J=8.5 Hz), 7.91 and 7.81 (2H, A2B2 each, J=8.5 Hz), 8.02 and ca. 7.94 (1H, br s each, NH), 12.71 (1H of enol form s, D2O exchangeable). 17: Colorless foam. HR-MS Calcd for C34H47NO6: 565.3401. Found: 565.3423. MS (m/z): 565 (M+, 27), 550 (17), 436 (11), 334 (10), 251 (15), 147 (20), 57 (100), 41 (27). IR (CHCl3) cm−1: 1711, 1667. 1H-NMR (CDCl3) δ: 1.27 (6H, s), 1.29 (6H, s), 1.30 (18H, s), 1.68 (4H, br s), 2.85 (2H, d, J=15 Hz), 2.91 (2H, d, J=15 Hz), 4.87 (1H, s, OH), 7.28 (1H, d, J=8.5 Hz), 7.43 (1H, dd, J=8.5, 2.5 Hz), 7.55 (2H, A2B2, J=8 Hz), 7.59 (1H, d, J=2.5 Hz), 7.86 (2H, A2B2, J=8 Hz), 7.98 (1H, br s, NH).
3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]-3-oxopropionic Acid (13′): CF3COOH (0.30 mL, 3.89 mmol) was added to a cooled (0°C) solution of 16 (42 mg, 93.5 µmol) in CH2Cl2 (2.7 mL) and the mixture was stirred at 0°C for 15 min and at an ambient temperature for 1.5 h. Volatile materials were evaporated off and the resulting crystalline residue was recrystallized to yield 13′ (31 mg, 84%) as colorless prisms, mp 183–184°C (CH2Cl2–hexane). MS (m/z): 349 (M+−CO2, 31), 334 (100), 292 (6), 147 (70), 119 (18), 104 (15), 91 (28), 76 (13), 43 (30). IR (KBr) cm−1: 1729, 1665, 1648. 1H-NMR of keto- and two enol-forms (ca. 12 : 5 : 3, DMSO-d6) δ: 1.23 (6H, s), 1.24 (6H, s), 1.64 (4H, s), 4.12 (2H of keto-form, s, D2O exchangeable), 5.74 and 5.96 (two OHs of enol-form, s each), 7.28 (1H, d, J=8 Hz), 7.57 (1H, dd, J=8, 2 Hz), 7.67 (1H, d, J=2 Hz), 7.92–8.11 (4H, m), 10.20 (NH, of an enol-form, br s), 10.26 (NH of keto- and the other enol-form, br s), 12.74, 12.99, and 13.43 (1H, br s each, COOH).
tert-Butyl 3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]-3-hydroxypropionate (18): NaBH4 (9 mg, 0.237 mmol) was added to a cooled (0°C) solution of 16 (36 mg, 80.2 µmol) in EtOH (3 mL), and the mixture was stirred at that temperature for 40 min. Quenching with satd. NH4Cl–H2O, extraction with EtOAc, usual work-up, and PTLC [hexane–EtOAc (5 : 2)] provided 18 (35 mg, 97%) as a colorless foam. HR-MS Calcd for C28H37NO4: 451.2721. Found: 451.2709. MS (m/z): 451 (M+, 53), 436 (72), 380 (39), 193 (63), 133 (34), 105 (34), 57 (100), 41 (39). IR (CHCl3) cm−1: 1701, 1665, 1607. 1H-NMR (CDCl3) δ: 1.27 (6H, s), 1.29 (6H, s), 1.45 (9H, s), 1.68 (4H, s), 2.64 (2H, d, J=6.5 Hz), 3.77 (1H, d, J=3 Hz, OH), 5.11 (1H, dt, J=3, 6.5 Hz), 7.29 (1H, d, J=8.5 Hz), 7.43 (2H, A2B2, J=8 Hz), 7.45 (1H, dd, J=8.5, 2 Hz), 7.55 (1H, d, J=2 Hz), 7.82 (2H, A2B2, J=8 Hz), 7.96 (1H, br s, NH).
3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]-3-hydroxypropionic Acid (12′): In the same manner as for the preparation of 13′ from 16, 18 (30 mg, 66.5 µmol) was treated with CF3COOH to afford 12′ (24 mg, 91%) as colorless prisms, mp 185–187°C (CH2Cl2–hexane). HR-MS Calcd for C24H29NO4: 395.2095. Found: 395.2071. MS (m/z): 395 (M+, 5), 380 (100), 362 (11), 320 (11), 193 (66), 133 (33), 105 (43), 77 (31), 43 (23). IR (KBr) cm−1: 1700, 1621, 1608. 1H-NMR (DMSO-d6) δ: 1.22 (6H, s), 1.23 (6H, s), 1.63 (4H, s), 2.53 (1H, dd, J=15, 8 Hz), 2.59 (1H, dd, J=15, 5.5 Hz), 5.01 (1H, dd, J=8, 5.5 Hz), 5.58 (1H, br s, OH), 7.26 (1H, d, J=8.5 Hz), 7.49 (2H, A2B2, J=8 Hz), 7.56 (1H, dd, J=8.5, 2 Hz), 7.67 (1H, d, J=2 Hz), 7.90 (2H, A2B2, J=8 Hz), 10.03 (1H, s), 12.16 (1H, br s, COOH).
Synthesis of 2 and 30(E)-3-(4-Carboxyphenyl)propenoic Acid (20): A slurry of 19 (120.0 g, 0.80 mol), malonic acid (124.8 g, 1.20 mol), and piperidine (8.0 mL, 81 mmol) in pyridine (350 mL) was heated with stirring at 80°C for 1.5 h. Then pyridine (50 mL) was added to this and the whole was further heated at 100°C for 2 h, and at reflux for 3 h. After having been cooled, the mixture was poured into an ice cooled HCl–H2O (6 N, 1.6 L), and precipitated crystals were collected by filtration under reduced pressure, washed with H2O, and thoroughly dried over P2O5 in vacuo to yield 20 (150.5 g, 98%) as slightly yellowish powder, mp >300°C (lit. mp 362–363°C, decomp15)). 1H-NMR (DMSO-d6) δ: 6.62 (1H, d, J=16 Hz), 7.61 (1H, d, J=16 Hz), 7.78 (A2B2, J=8.5 Hz), 7.94 (A2B2, J=8.5 Hz).
3-(4-Carboxyphenyl)propionic Acid (21): The acid 20 (50.0 g, 0.26 mol) was added in small portions to a solution of Na2CO3 (31.8 g, 0.30 mol) in H2O (250 mL) in a Parr vessel (500 mL) with stirring. Pd/C (10%, 750 mg) was added to this and the resulting slurry was hydrogenated under H2 atmosphere (0.3 MPa) for 70 h. The mixture was filtered through a Celite bed and the Celite was rinsed with H2O. The filtrate and wash water were combined and conc. HCl–H2O was added dropwise to adjust pH to ca. 2. Precipitated crystals were collected as above to give 21 (47.9 g, 95%) as a colorless powder, mp 286–290°C [lit. mp 286–289°C (HOAc)19)]. IR (KBr) cm−1: 1693, 1681. 1H-NMR (DMSO-d6) δ: 2.55 (2H, t, J=7.5 Hz), 2.87 (2H, t, J=7.5 Hz), 7.33 (A2B2, J=8 Hz), 7.83 (A2B2, J=8 Hz).
3-(4-Carboxyphenyl)propionic Acid-2,2,3,3-d4 (22): Palladium-carbon (10%, 1.50 g) was added to a slightly slurried solution of 21 (30.0 g, 0.155 mol), Na2CO3 (16.4 g, 0.155 mol) in D2O (99%D, 300 mL) in a vessel (500 mL, Ace Glass, globular shape), and it was purged with hydrogen gas (1 atm) and sealed, and then heated in an oil bath (130°C) for 64 h. After termination of the reaction (checked by NMR of the aliquot in D2O), the mixture was filtered through a Celite bed under reduced pressure, and the filtrate was distilled to recover D2O (247 mL, 82% at 57–58°C/120 mmHg). The resulting residue and wash water of the Celite bed were combined, and the solution was made acidic (pH ca. 2) with conc. HCl–H2O. Precipitated crystals were collected by filtration under reduced pressure, washed with water, and then thoroughly dried over P2O5 in vacuo to provide desired 22 (29.8 g, 97%, D ratio of X=97–98%, Y=98%) as a colorless powder, mp 271–273°C. HR-MS: Calcd for C10H6D4O4: 198.0830. Found: 198.0825. MS (m/z): 198 (M+, 67), 181 (11), 152 (50), 137 (61), 109 (100), 93 (19), 79 (25), 45 (28). IR (KBr) cm−1: 1683. 1H-NMR (1% Na2CO3–D2O) δ: 2.35 (0.04–0.06H, br s), 2.79 (0.04H, br s), 7.22 (A2B2, J=8 Hz), 7.68 (A2B2, J=8 Hz).
3-(4-Carboxyphenyl)propionic Acid-3,3-d2 (23): Palladium-carbon (10%, 250 mg) was added to a slightly slurried solution of 21 (5.00 g, 25.8 mmol), Na2CO3 (3.28 g, 30.9 mmol) in D2O (99%D, 100 mL) in a Parr vessel (500 mL, circular cylinder), and it was purged with hydrogen gas (1 atm) and sealed, and then heated at 100°C for 48 h. After termination of the reaction (NMR check as above), the mixture was treated in the same manner as above to yield 23 (4.90 g, 97%, D ratio of X=0%, Y>98%) along with recovered D2O (84 mL, 84%). 23: Colorless powder, mp 273–276°C. HR-MS: Calcd for C10H8D2O4: 196.0704. Found: 196.0698. MS (m/z): 196 (M+, 70), 179 (8), 150 (49), 137 (64), 109 (100), 106 (31), 93 (23), 78 (33), 45 (30). IR (KBr) cm−1: 1697, 1683. 1H-NMR (1% Na2CO3–D2O) δ: 2.38 (2H, s), ca. 2.77–2.83 (<0.04H, m), 7.23 (A2B2, J=8 Hz), 7.69 (A2B2, J=8 Hz).
Methyl 3-(4-Carboxyphenyl)propionate-2,2,3,3-d4 (24) and Diester 25: SOCl2 (14 µL, 0.192 mmol) was added to a cooled (0°C) slurry of 22 (1.93 g, 9.75 mmol) in MeOH (30 mL) and the mixture was stirred at an ambient temperature for 16 h. The resulting clear solution was made slightly acidic (pH 6) by addition of satd. NaHCO3–H2O (0.8 mL) and the solvent was evaporated off. Et2O was added to the residue and the mixture was extracted with Na2CO3–H2O (10% w/v). The Et2O layer was washed with satd. NH4Cl–H2O and then treated as usual. Purification by PTLC [hexane–EtOAc (5 : 1)] gave 25 (118 mg, 5%). On the other hand, conc. HCl–H2O was added dropwise to the cooled (0°C) basic water layer to adjust pH to ca. 2, and a precipitated mixture of 24 and recovered 22 was collected and dried as above. The mixture was dissolved in CH2Cl2 to make a slight slurry, which was filtered through a filter paper to recover 22 (54 mg, 3%). The filtrate was recrystallized to yield 24 (1.80 g, 87%) as colorless scales, mp 149–150.5°C (CH2Cl2–hexane). HR-MS: Calcd for C11H8D4O4: 212.0986. Found: 212.1001. MS (m/z): 212 (M+, 34), 195 (4), 181 (12), 152 (100), 137 (40), 134 (18), 123 (15), 109 (76), 79 (16). IR (KBr) cm−1: 1719, 1681. 1H-NMR (CDCl3) δ: 2.65 (0.02–0.03H, br s), 3.01 (0.02H, br s), 3.67 (3H, s), 7.32 (A2B2, J=8.5 Hz), 8.04 (A2B2, J=8.5 Hz). 13C-NMR (CDCl3) δ: 30.2 (quint, JCD=20 Hz), 34.4 (quint, JCD=20.5 Hz), 51.7, 127.5, 128.5, 130.5, 146.8, 172.0, 173.0. 25: colorless oil. HR-MS: Calcd for C12H10D4O4: 226.1142. Found: 226.1146. MS (m/z): 226 (M+, 47), 195 (69), 166 (100), 151 (50), 134 (46), 123 (47), 59 (26). IR (neat) cm−1: 1727, 1714. 1H-NMR (CDCl3) δ: 2.60–2.64 (0.02–0.03H, m), ca. 2.98 (0.02H, br s), 3.67 (3H, s), 3.90 (3H, s), 7.27 (A2B2, J=8.5 Hz), 7.96 (A2B2, J=8.5 Hz).
Methyl 3-(4-Carboxyphenyl)propionate-3,3-d2 (26) and Diester 27: In the same manner as for the preparation of 24 and 25, 23 (2.84 g, 14.5 mmol) was treated with SOCl2 (21 µL, 0.29 mmol) in MeOH (40 mL) to provide 26 (2.61 g, 86%), 27 (0.21 g, 6%) along with recovered 23 (23 mg, 1%). 26: Colorless scales, mp 149–150°C (CH2Cl2–hexane). HR-MS: Calcd for C11H10D2O4: 210.0860. Found: 210.0844. MS (m/z): 210 (M+, 36), 179 (13), 150 (100), 137 (42), 132 (15), 123 (14), 109 (21), 107 (18), 78 (19). IR (KBr) cm−1: 1720, 1681. 1H-NMR (CDCl3) δ: 2.66 (2H, s), ca. 2.98–3.04 (ca. 0.03H, m), 3.68 (3H, s), 7.31 (A2B2, J=8.5 Hz), 8.03 (A2B2, J=8.5 Hz). 27: Colorless oil. HR-MS: Calcd for C12H12D2O4: 224.1017. Found: 224.1004. MS (m/z): 224 (M+, 49), 193 (66), 164 (100), 151 (39), 132 (39), 123 (43), 105 (20), 92 (20), 59 (24). IR (neat) cm−1: 1730 (sh), 1715. 1H-NMR (CDCl3) δ: 2.64 (2H, s), ca. 2.95–3.02 (0.03H, m), 3.67 (3H, s), 3.90 (3H, s), 7.27 (A2B2, J=8.5 Hz), 7.96 (A2B2, J=8.5 Hz).
Methyl 3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propionate-2,2,3,3-d4 (28): Different from the case of the preparation of 5, the yield of 28 was calculated not from 4, but from the time-consuming compound 24. SOCl2 (10.3 mL, 141 mmol) and N,N-dimethylformamide (DMF) (0.15 mL, 1.94 mmol) were added to a slurry of 24 (10.00 g, 47.2 mmol) in benzene (75 mL), and the mixture was refluxed with stirring for 6 h. The volatile materials were evaporated off, then benzene (40 mL) was added to the resulting residue and evaporated again to dryness. The residue was dissolved in CH2Cl2 (80 mL) and the solution was cooled in an ice bath. A solution of 4 (10.06 g, 49.6 mmol) and Et3N (19.7 mL, 142 mmol) in CH2Cl2 (20 mL) was added to this and the mixture was stirred at that temperature for 30 min, and at an ambient temperature for 15 h. Saturated NaHCO3–H2O was added and the whole was extracted with CHCl3. Usual work-up followed by purification by recrystallization and column chromatography [hexane–EtOAc (4 : 1)] gave 28 (18.06 g, 96%) as colorless prisms, mp 136.5–137.5°C (CH2Cl2–hexane). The H2O layer was made acidic to pH ca. 2 with 10% HCl–H2O and extracted with EtOAc. Usual work-up and recrystallization recovered 24 (85 mg, 1%). HR-MS: Calcd for C25H27D4NO3: 397.2553. Found: 397.2569. MS (m/z): 397 (M+, 36), 382 (100), 366 (4), 195 (65), 137 (12), 134 (16), 108 (8), 107 (8). IR (KBr) cm−1: 1708, 1659. 1H-NMR (CDCl3) δ: 1.28 (6H, s), 1.30 (6H, s), 1.69 (4H, s), 2.64 (0.03–0.04H, br s), 2.99 (0.02–0.03H, br s), 3.68 (3H, s), 7.30 (1H, d, J=8.5 Hz), 7.31 (A2B2, J=8 Hz), 7.41 (1H, dd, J=8.5, 2 Hz), 7.53 (1H, d, J=2 Hz), 7.71 (1H, br s, NH), 7.80 (A2B2, J=8 Hz).
Methyl 3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propionate-3,3-d4 (29): In the same manner as for the preparation of 28, 26 (2.402 g, 11.4 mmol) was condensed with 3 (2.438 g, 12.0 mmol) to form 29 (4.364 g, 97%) as colorless prisms, mp 135–136°C (CH2Cl2–hexane). From the H2O layer, 26 (33 mg, 1%) was recovered as above. HR-MS: Calcd for C25H29D2NO3: 395.2428. Found: 395.2430. MS (m/z): 395 (M+, 37), 380 (100), 193 (68), 132 (19), 105 (14), 43 (8). IR (KBr) cm−1: 1709, 1660, 1608. 1H-NMR (CDCl3) δ: 1.28 (6H, s), 1.30 (6H, s), 1.69 (4H, s), 2.65 (2H, s), 2.97–3.04 (ca. 0.03H, m), 3.68 (3H, s), 7.30 (1H, d, J=8.5 Hz), 7.32 (A2B2, J=8 Hz), 7.41 (1H, dd, J=8.5, 2.5 Hz), 7.52 (1H, d, J=2.5 Hz), 7.39 (1H, br s, NH), 7.80 (A2B2, J=8 Hz).
3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propionic Acid-2,2,3,3-d4 (IT-YA-00616, 2): In the same manner as for the preparation of 1 from 5, 28 (10.25 g, 25.8 mmol) was hydrolyzed with LiOH·H2O (1.30 g, 31.0 mmol) to afford 2 (9.70 g, 98%) as colorless fine needles, mp 230.5–231.5°C (EtOAc). Anal. Calcd for C24H25D4NO3: C, 75.16; H(+D), 7.62; N, 3.65. Found: C, 75.00; H(+D), 7.57; N, 3.64. HR-MS: Calcd for C24H25D4NO3: 383.2397. Found: 383.2395. MS (m/z): 383 (M+, 37), 368 (100), 326 (2), 189 (6), 181 (73), 109 (19), 107 (8), 43 (6). IR (KBr) cm−1: 1707, 1615. 1H-NMR (CDCl3) δ: 1.27 (6H, s), 1.30 (6H, s), 1.69 (4H, s), 2.69 (ca. 0.04H, br s), 3.01 (ca. 0.03H, br s), 7.30 (1H, d, J=8.5 Hz), 7.33 (A2B2, J=8 Hz), 7.42 (1H, dd, J=8.5, 2 Hz), 7.53 (1H, d, J=2 Hz), 7.76 (1H, br s, NH), 7.80 (A2B2, J=8 Hz). 13C-NMR (DMSO-d6) δ: 31.62, 31.64, 33.5, 34.0, 34.58, 34.63, 118.0, 118.2, 126.3, 127.6, 128.2, 132.8, 136.7, 139.7, 144.46, 144.54, 165.1, 173.6. The 13C-NMR spectrum was measured in DMSO-d6 in order to obtain a sufficiently high concentration of 2, but the signals ascribed to two CD2 carbons were observed only as broadened weak peaks at somewhat higher field (ca. 29.4 and 34.2 ppm) than those of 1 (30.2 and 34.9 ppm).
3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propionic Acid-3,3-d2 (30): In the same manner as for the preparation of 1 from 5, 29 (4.05 g, 10.3 mmol) was hydrolyzed with LiOH·H2O (0.52 g, 12.4 mmol) to afford 30 (3.84 g, 98%) as colorless fine needles, mp 229.5–230°C (EtOAc). HR-MS: Calcd for C24H27D2NO3: 381.2271. Found: 381.2253. MS (m/z): 381 (M+, 33), 366 (100), 149 (90), 109 (27), 105 (19), 79 (11). IR (KBr) cm−1: 1707, 1616. 1H-NMR (CDCl3) δ: 1.28 (6H, s), 1.30 (6H, s), 1.69 (4H, s), 2.71 (2H, s), ca. 2.99–3.04 (ca. 0.03H, m), 7.30 (1H, d, J=8.5 Hz), 7.33 (A2B2, J=8.5 Hz), 7.41 (1H, dd, J=8.5, 2 Hz), 7.53 (1H, d, J=2 Hz), 7.74 (1H, br s, NH), 7.80 (A2B2, J=8.5 Hz).
Preparation of the Congener 325-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]benzyl]-2,2-dimethyl-1,3-dioxane-4,6-dione (31): Meldrum’s acid (59 mg, 0.409 mmol), diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate (Hantzsch ester, 99 mg, 0.392 mmol) and L-proline (9 mg, 78.2 µmol) were added in this order to a slurry of 1 (125 mg, 0.373 mmol) in CH3CN (3 mL) and the mixture was stirred at an ambient temperature for 22 h. At this point, the mixture became nearly clear. The solvent was evaporated off and the resulting residue was subjected to column chromatography [hexane–EtOAc (2 : 1)] to yield 31 (146 mg, 85%) as colorless fine needles, mp 145–146°C (decomp. CH2Cl2–hexane). MS (m/z): 379 (M+−C4H4O2, 31, converted to 1 in the MS chamber), 364 (100), 177 (67), 107 (18), 57 (25), 43 (25). IR (KBr) cm−1: 1780, 1740, 1641. 1H-NMR (CDCl3) δ: 1.27 (6H, s), 1.30 (6H, s), 1.61 (3H, s), 1.69 (4H, s), 1.77 (3H, s), 3.54 (2H, d, J=5 Hz), 3.80 (1H, t, J=5 Hz), 7.30 (1H, d, J=8.5 Hz), 7.41 (1H, dd, J=8.5, 2.5 Hz), 7.44 (A2B2, J=8.5 Hz), 7.53 (1H, d, J=2.5 Hz), 7.76 (1H, br s, NH), 7.79 (A2B2, J=8.5 Hz).
3-[4-[(5,6,7,8-Tetrahydro-5,5,8,8-tetramethyl-2-naphthyl)carbamoyl]phenyl]propionic Acid-2,2-d2 (32): D2O (0.5 mL) was added to a solution of 31 (145 mg, 0.313 mmol) in pyridine (5 mL) and the mixture was stirred under reflux for 16 h. After cooling, HCl–H2O (2 N) was added and the whole was extracted with CHCl3. Usual work-up and recrystallization of the residue provided 32 (96 mg, 80%) as colorless scales, mp 226.5–227.5°C (EtOAc–hexane). HR-MS Calcd for C24H27D2NO3: 381.2271. Found: 381.2256. MS (m/z): 381 (M+, 38), 366 (100), 179 (78), 107 (17), 105 (11). IR (KBr) cm−1: 1712, 1619. 1H-NMR (CDCl3) δ: 1.28 (6H, s), 1.30 (6H, s), 1.69 (4H, s), 3.03 (2H, br s), 7.30 (1H, d, J=8.5 Hz), 7.33 (A2B2, J=8.5 Hz), 7.42 (1H, dd, J=8.5, 2 Hz), 7.53 (1H, d, J=2 Hz), 7.73 (1H, br s, NH), 7.81 (A2B2, J=8.5 Hz).