Regular Articles
Synthesis of New Nicotinic Acid Derivatives and Their Evaluation as Analgesic and Anti-inflammatory Agents
2013 年 61 巻 9 号 p. 933-940
詳細
2013 年 61 巻 9 号 p. 933-940
A series of 2-substituted phenyl derivatives of nicotinic acid 4a–l were synthesized and evaluated for their analgesic and anti-inflammatory activities. Compounds including 2-bromophenyl substituent, 4a, c, and d, proved to display distinctive analgesic and anti-inflammatory activities in comparison to mefenamic acid as a reference drug. Compound 4c could be identified as the most biologically active member within this study with an interesting dual anti-inflammatory analgesic profile. Effect of the compounds 4a–l on the serum level of certain inflammatory cytokines such as tumor necrosis factor (TNF)-α and interleukin (IL)-6 was also determined.
Osteoarthritis (OA) is the most common cause of chronic joint pain. Degeneration of joint cartilage is the central feature in OA, but the disease is associated with concomitant changes in synovium and subchondral bone metabolism, causing inflammation of the synovial membrane in the involved joints.1) One of the factors that stimulate OA, results from the failure of chondrocytes that lie within the joint to synthesize a good-quality matrix, and to maintain a balance between synthesis and degradation of the extracellular matrix. Thus, synovial inflammation and local concentration of pro-inflammatory mediators seems to be directly involved in the generation of pain in OA joints.2)
Recent studies pointed out the key roles of inflammatory cytokines, such as tumor necrosis factor (TNF)-α and interleukin (IL)-6, in the etiology and pathogenesis of rheumatoid arthritis (RA) and osteoarthritis.3,4) Thus, therapies suppressing these inflammatory cytokines have been focused on, as an effective treatment of these inflammatory diseases. In addition, myeloperoxidase (MPO) plays an important role in infection and inflammation through converting hydrogen peroxide and chloride to hypochlorous acid and water.5) The excessive MPO activity, and production of reactive oxygen species (ROS), especially HOCl, are implicated in many inflammatory processes, including atherosclerosis, cancer, and Alzheimer’s disease.6,7) MPO is released in the extracellular medium by highly stimulated and dying neutrophils in pathological conditions of acute and chronic inflammation. Under these conditions, MPO is able to exert oxidant activity on neighbouring cells and tissues.8)
The medications most commonly used to treat OA are symptom-modifying drugs, such as nonsteroidal anti-inflammatory drugs (NSAIDs), which inhibit prostaglandin (PG) formation through inhibition of both cyclooxygenase (COX)-1 and COX-2 enzymes. However, the chronic use of currently available NSAIDs may elicit appreciable gastrointestinal (GI) toxicity leading to ulceration, bleeding and nephrotoxicity, which causes some patients to abandon NSAID therapy.9–11)
On this basis, synthetic approaches based upon chemical modification of classic NSAIDs have been taken with the aim of improving their safety profile. Pyridine derivatives form an important class of heterocyclic compounds that have attracted the attention of many scientists. The pyridine nucleus is present in niflumic acid and flunixin, which are a traditional NSAIDs belonging to the class of fenamates (Fig. 1). Unlike most other NSAIDs, the fenamates appear also to compete with PGs for binding at the PG receptor site and, thus, potentially antagonize the physiopathological effects of PGs that have already been formed.12) However, fenamates are still exhibit most of the GI adverse effects induced by NSAIDs. GI damage from NSAIDs may be generally attributed to the direct contact of the carboxylic acid (–COOH) moiety of NSAIDs with GI mucosal cells (topical effect) as well as decreased tissue PG production in tissues thus reducing the cytoprotective effect of PGs in maintaining GI health.13)

Several studies have described the derivatization of the carboxylic function with amide or N-acylarylhydrazone moieties.14–17) N-Acylhydrazones have been widely described as potent anti-inflammatory, antinociceptive, and anti-platelet compounds, due to their ability to mimic the bis-allylic moiety of unsaturated fatty acids and amides, such as arachidonic acid.14)
In light of the above facts, this work comprises the synthesis and evaluation of analgesic and anti-inflammatory activities of new series of N-acylarylhydrazone derivatives of 2-arylaminonicotinic acids and 2-aryloxynicotinic acids (Fig. 1).
The synthetic route to the target compounds 4a–l is outlined in Chart 1. Briefly, the 2-substituted nicotinic acids 1a–c, obtained according to a procedure previously described,18) were esterified to give the corresponding ethyl nicotinate derivatives 2a–c. The appearance of signals in 1H-NMR spectra, at δ 1.28–1.35 ppm and 4.25–4.40 ppm assignable to CH3 and CH2 protons respectively of –COOCH2CH3 confirms the formation of ester 2. The latter compounds were also confirmed by their IR spectra, which revealed a band at 1735–1750 cm−1 indicating ester carbonyl function.
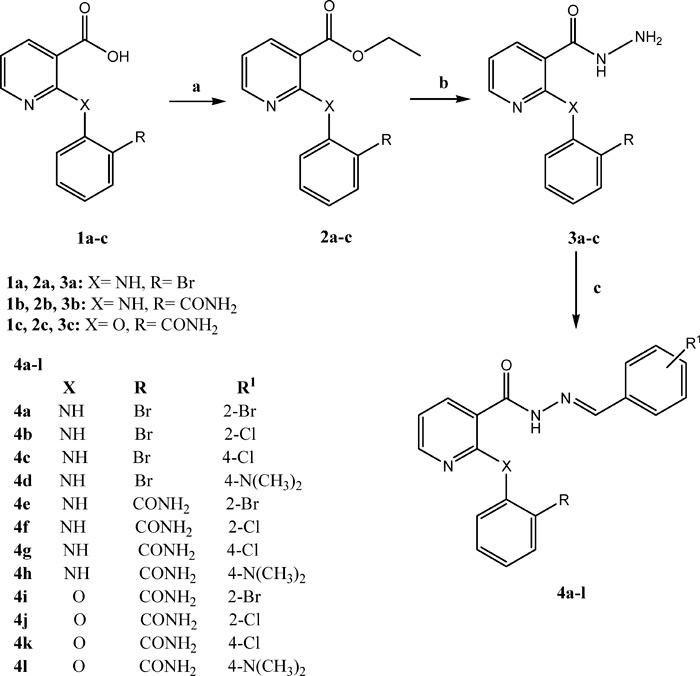
Reagents and conditions: a: Abs. C2H5OH, conc. H2SO4, 8 h, b: N2H4·H2O, C2H5OH, 3 h, c: ArCHO, C2H5OH, 4 h.
Reaction of 2a–c with hydrazine hydrate in absolute ethanol, afforded the corresponding hydrazides 3a–c, which are key intermediates for the production of the title compounds 4a–l. IR spectra of 3a–c, showed bands due to carbonyl amide function in the range of 1639–1697 cm−1. The target hydrazone compounds 4a–l were prepared in good yields by the condensation of the hydrazides 3 with different substituted aromatic aldehydes in refluxing ethanol. The compounds containing arylidene–hydrazide structure may exist as E/Z geometrical isomers about –C=N– double bond and cis/trans amide conformers.19) According to literature, the presence of single downfield resonating (8.43–8.99 ppm) imine hydrogen signal accounts for formation of E-isomers exclusively, in DMSO-d6 solution, whereas Z-isomer can be stabilized in less polar solvents by an intramolecular hydrogen bond.20) In the present study, the spectral data were recorded in DMSO-d6, and the signal corresponding to the imine hydrogen was observed in the range of 8.19–8.93 ppm.
Analytical and spectral data (IR, 1H-NMR, 13C-NMR, mass spectra) of the synthesized compounds were in full agreement with the proposed structures.
Analgesic ActivityIn this study, the new compounds were tested for analgesic activity using the writhing test in mice21) and compared to mefenamic acid, as a reference standard at the same dose level (25 mg/kg, per os (p.o.)). Writhing was induced by intraperitoneal (i.p.) injection of freshly prepared acetic acid solution (1%, 10 mL/kg) in mice. The number of writhes (constriction of abdomen, turning of trunk, extension of hind limbs) due to acetic acid was expressed as a nociceptive response. Vehicle treated control mice were given acetic acid solution (1%) and writhing response was noted during the period of 15 min. The results showed that the tested compounds exhibited analgesic activity ranging from 22.2±1.2% to 73.7±6.2%. Compounds 4a, c, and d showed significant reduction in the writhing response (61.7±4.8 to 73.7±6.2). Compound 4c showed analgesic effect similar to that exhibited by mefenamic acid (72.4±4.6%). The compounds that displayed the highest activity in the writhing test include 2-bromo substituent in the phenyl ring. Replacement of the 2-bromo substituent by carboxamide moiety results in significant reduction in activity (compounds 4e–h). It was also obvious that, among the compounds bearing carboxamide substituent in the 2-position of the phenyl ring (compounds 4e–l), higher analgesic activity was observed with the aryloxy analogues (X=O), (compounds 4i–l) (Table 1).
| Compound | Analgesic activity (% protection) | Anti-inflammatory activity (% inhibition) | Ulcerogenic potential (Severity index) |
|---|---|---|---|
| Vehicle | 0 | 0 | 0 |
| Mefenamic acid | 72.4±4.6* | 59.3±4.21* | 1.0±0.06* |
| 4a | 61.7±4.8* | 52.9±3.8*,# | 0.82±0.05*,# |
| 4b | 54.8±4.2*,# | 39.2±2.5*,# | 0.72±0.05*,# |
| 4c | 73.7±6.2* | 62.2±5.4* | 1.3±0.09*,# |
| 4d | 70.5±4.8* | 56.3±4.2* | 0.86±0.05* |
| 4e | 25.4±2.1*,# | 15.5±0.8*,# | 0.5±0.02*,# |
| 4f | 28.2±1.1*,# | 22.3±1.2*,# | 0.3±0.01*,# |
| 4g | 22.2±1.2*,# | 23.1±0.62*,# | 0.41±0.02*,# |
| 4h | 55.2±4.3*,# | 32.1±1.8*,# | 0.69±0.05*,# |
| 4i | 24.8±2.3*,# | 12.0±0.9*,# | 0.2±0.01*,# |
| 4j | 52.3±0.42*,# | 41.0±3.4*,# | 0.64±0.03*,# |
| 4k | 42.1±0.38*,# | 31.2±0.16*,# | 0.18±0.01*,# |
| 4l | 52.3±0.42*,# | 22.1±0.18*,# | 0.22±0.01*,# |
The vehicle group was pretreated with 1% carboxymethylcellulose solution. All compounds were tested in a dose equals 25 mg/kg (p.o.) 30 min before testing. Data are presented as mean±S.E.M. and analyzed using one-way ANOVA followed by Tukey’s test for multiple comparisons. * p<0.05 compared to the vehicle group and # p<0.05 compared to mefenamic acid group.
Anti-inflammatory activity was determined by using carrageenan induced rat paw edema model.22) Carrageenan (1% w/v) was used to produce paw edema. Edema is presented as percentage increase in right hind paw, in comparison to the uninjected left hind paw. Percentage change in paw volume was calculated and expressed as the amount of inflammation. Compounds 4a, c, and d significantly induced strong anti-inflammatory effect that was comparable to that exhibited by mefenamic acid. These results were in agreement with those shown in the analgesic activity test (Table 1).
Furthermore, mefenamic acid as well as the tested compounds 4a–d, have shown significant suppression of serum IL-6 and TNF-α levels compared to the control group (Table 2).
| Compound | TNF-α (mg/mL) | IL-6 (mg/mL) |
|---|---|---|
| Vehicle | 5.24±0.42 | 20.4±1.6 |
| Mefenamic acid | 3.04±0.27* | 8.2±0.54* |
| 4a | 3.32±0.28* | 10.7±0.87*,# |
| 4b | 3.55±0.32* | 14.7±0.12*,# |
| 4c | 2.67±0.21* | 8.3±0.65* |
| 4d | 3.12±0.27* | 9.6±0.76* |
| 4e | 4.38±0.42*,# | 16.5±1.22,# |
| 4f | 4.23±0.36*,# | 16.8±1.31,# |
| 4g | 4.78±0.45,# | 17.7±1.42,# |
| 4h | 3.42±0.31*,# | 12.2±1.16*,# |
| 4i | 4.54±0.43,# | 17.1±0.12,# |
| 4j | 3.72±0.31*,# | 13.5±0.11* ,# |
| 4k | 5.12±0.47,# | 21.34±0.24,# |
| 4l | 4.98±0.45,# | 22.34±0.21,# |
All compounds were tested in a dose equals 25 mg/kg (p.o.) 30 min before testing. Data are represented as mean±S.E.M. and analyzed using one-way ANOVA followed by Tukey’s test for multiple comparisons. * p<0.05 compared to the vehicle group and # p<0.05 compared to mefenamic acid group.
In current study, testing acute ulcerogenic potential revealed that mefenamic acid showed an ulcer severity index of 1.0±0.06. All tested compounds except 4c showed lower mean severity index than that observed by mefenamic acid, indicating lower ulcerogenic effect (Table 1). Although compound 4c revealed promising analgesic and anti-inflammatory results, it displayed higher severity index than mefenamic acid. The results of ulcerogenicity studies revealed that derivatization of the carboxylic acid group slightly improved the safety margin of the all final new compounds with the exception of 4c. Thus, the ulcerogenic effects of the compounds 4a–l may be attributed to the non-selective suppression of cyclooxygenases COX-1 and COX-2 isoforms, thereby preventing the biosynthesis of PGs that have a role in homeostasis and gastroprotection.
Immunohistochemistry for Myeloperoxidase EnzymeMPO is released in the extracellular medium by highly stimulated and dying neutrophils in pathological conditions of acute and chronic inflammation. Under these conditions, MPO is able to exert oxidant activity on neighbouring cells and tissues. Compounds that possess antioxidant activity have the ability to inhibit MPO level, and thus suppress oxidative damage and may consequently, prevent inflammatory conditions.23)
Histopathological examination revealed that the mucosa of mefenamic acid group as well as groups treated with compounds 4a–e, h, and j showed higher staining for myeloperoxidase enzyme compared to the vehicle group (Figs. 2A, B).

Data are expressed as mean±S.E.M. and analyzed using one-way ANOVA followed by Bonferroni’s post-hoc test at p<0.05. *p<0.05 compared to the vehicle group and #p<0.05 compared to mefanamic acid group.
Various 2-substitutedphenyl derivatives of nicotinic acid 4a–l structurally related to niflumic acid were synthesized and screened for their potential analgesic, anti-inflammatory and ulcerogenic potentials. Promising analgesic and anti-inflammatory activities compared to the reference drug, mefenamic acid, was observed with the compounds 4a, c, and d. It has been also obvious that the latter compounds significantly reduced serum TNF-α and IL-6 levels.
The tested compounds 4a–l were also screened for ulcerogenic adverse effect. The results revealed that all compounds except 4c showed lower ulcerogenic potential than the reference drug.
From close inspection of the results of in vivo experiments in this study and our previous investigation in this field,18) it is obvious that derivatization of carboxylic acid functionality with N-acyl hydrazone in compounds 4a, c, and d resulted in significant increase in both analgesic and anti-inflammatory activities, without affecting the ulcerogenic potential compared to the corresponding analogue with free carboxylic acid grouping. On the other hand, derivatization of the carboxylic acid function suppress the analgesic and anti-inflammatory properties of compounds 4e–4h with concomitant suppression of ulcerogenicity, compared to the free carboxylic acid analogues.
In light of these results, one can say that the most promising results as the anti-inflammatory and analgesic agents were observed with the nicotinic acid derivatives 4a, c, and d, containing 2-bromophenyl substituent in their structure.
All chemicals and reagents were obtained from Aldrich (Sigma-Aldrich, St. Louis, MO, U.S.A.), and were used without further purification. Reactions were monitored by TLC, performed on silica gel glass plates containing 60 GF-254, and visualization on TLC was achieved by UV light or iodine indicator. IR spectra were determined on Shimadzu IR 435 spectrophotometer (KBr, cm−1). 1H-NMR spectra were carried out using a Mercury 300-BB 300 MHz using tetramethylsilane (TMS) as internal standard. 13C-NMR spectra were carried out using a Mercury 300-BB 75 MHz using TMS as internal standard. Chemical shifts (δ) are recorded in ppm on δ scale, Micro Analytical Center, Cairo University, Egypt. Mass spectra were recorded on Shimadzu Qp-2010 plus Spectrometer, Micro Analytical Center, Cairo University, Egypt. Elemental analyses were carried out at the Micro Analytical Center, Cairo University, Egypt. Melting points were determined with Stuart apparatus and are uncorrected. Progress of the reactions was monitored using TLC sheets precoated with UV fluorescent silica gel Merck 60F 254 using acetone–benzene (1 : 9) and were visualized using UV lamp.
The starting materials 2-(2-bromophenylamino)nicotinic acid 1a, 2-(2-carbamoylphenylamino)nicotinic acid 1b and 2-(2-carbamoylphenoxy)nicotinic acid 1c were prepared as reported.18) Other chemicals were obtained from Aldrich, Fluka, or Merck Chemicals.
General Procedure for the Preparation of 2a–cTo a solution of an appropriate 1a–c (10 mmol) in absolute ethanol (30 mL), conc H2SO4 (1 mL) was added, and the mixture was heated under reflux for 8 h. After cooling, the reaction mixture was concentrated under reduced pressure, an aqueous sodium carbonate solution (20 mL, 10%, w/v) was added, and the ester was extracted with chloroform (2×15 mL). The combined extract was dried (Na2SO4), filtered, concentrated under reduced pressure, and the resulting ester was collected.
Ethyl 2-(2-Bromophenylamino)nicotinate (2a): Yield 68%; mp 41–42°C; IR (KBr) cm−1: 3429 (NH), 3055 (C–H aromatic), 2981, 2830 (C–H aliphatic), 1740 (C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 1.32 (t, 3H, J=7.2 Hz, CH3), 4.33 (q, 2H, J=7.2 Hz, CH2), 5.21 (s, 1H, NH, D2O exchangeable), 6.78–7.66 (m, 5H, 4H Ar and H5, pyr.), 8.25 (d, 1H, J=6.2 Hz, H4, pyr.), 8.66 (d, 1H, J=5.4 Hz, H6, pyr.); MS (electron ionization (EI)) m/z (% rel. Int.): 322 (M+2, 1.74), 320 (M+, 1.88); Anal. Calcd for C14H13BrN2O2: C, 52.36; H, 4.08; N, 8.72. Found: C, 52.11; H, 3.98; N, 8.45.
Ethyl 2-(2-Carbamoylphenylamino)nicotinate (2b): Yield 60%; mp 104–105°C; IR (KBr) cm−1: 3500–3200 (NH, NH2), 3124 (C–H aromatic), 2985, 2850 (C–H aliphatic), 1645, 1735 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 1.35 (t, 3H, J=7.5 Hz, CH3), 4.40 (q, 2H, J=7.5 Hz, CH2), 7.04–7.09 (m, 1H, Ar), 7.52–7.55 (m, 1H, Ar), 7.70 (d, 1H, Ar), 7.88–7.94 (m, 2H, 1H Ar, H5, pyr.), 8.25 (d, 1H, J=6.0 Hz, H4, pyr.), 8.88 (d, 1H, J=5.1 Hz, H6, pyr.), 11.44 (s, 2H, NH2, D2O exchangeable); MS (EI) m/z (% rel. Int.): 286 (M+H, 0.16), 285 (M+, 0.47), 241 (C14H13N2O2, 64.46); Anal. Calcd for C15H15N3O3: C, 63.15; H, 5.30; N, 14.73. Found: C, 63.23; H, 5.05; N, 14.43.
Ethyl 2-(2-Carbamoylphenoxy)nicotinate (2c): Yield 55%; mp 45–46°C; IR (KBr) cm−1: 3429–3380 (NH, NH2), 3065 (C–H aromatic), 2950, 2845 (C–H aliphatic), 1640, 1750 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 1.28 (t, 3H, J=7.2 Hz, CH3), 4.25 (q, 2H, J=7.2 Hz, CH2), 6.81–6.87 (m, 1H, Ar), 7.33–7.41 (m, 1H, Ar), 7.52–7.58 (m, 1H, Ar), 7.83–7.85 (m, 2H, 1H Ar, H5, pyr.), 8.23 (d, 1H, J=7.8 Hz, H4, pyr.), 8.56 (d, 1H, J=4.8 Hz, H6, pyr.), 10.55 (s, 2H, NH2, D2O exchangeable); MS (EI) m/z (% rel. Int.): 285 (M−H, 0.22); Anal. Calcd for C15H14N2O4: C, 62.93; H, 4.93; N, 9.79. Found: C, 62.78; H, 5.11; N, 9.44.
General Procedure for the Preparation of 3a–cA mixture of the appropriate 2a–c (1 mmol) and hydrazine hydrate (80%, 5 mmol) in ethanol, was heated under reflux for 3 h. The reaction mixture was concentrated under reduced pressure and left overnight. The resulting crystalline solid product was filtered then washed with ethanol to give pure acid hydrazides 3a–c.
2-(2-Bromophenylamino)nicotinohydrazide (3a): Yield 74%; mp 199–200°C; IR (KBr) cm−1: 3433–3201 (NH, NH2), 3124 (C–H aromatic), 1689 (C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 6.46–6.60 (m, 3H, 2H Ar, H5, pyr.), 7.68–7.71 (d, 2H, 1H Ar, H4, pyr.), 8.40 (s, 1H, NH, D2O exchangeable), 8.28–8.31 (m, 2H, 1H Ar, H6, pyr.), 10.54 (s, 3H, NH, NH2, D2O exchangeable); MS (EI) m/z (% rel. Int.): 308 (M+2, 4.64), 306 (M+, 4.91); Anal. Calcd for C12H11BrN4O: C, 46.93; H, 3.61; N, 18.24. Found: C, 46.88; H, 3.55; N, 18.46.
2-[(3-Hydrazinecarbonyl)pyridine-2-ylamino]benzamide (3b): Yield 60%; mp 219–220°C; IR (KBr) cm−1: 3444–3302 (NH, NH2), 3078 (C–H aromatic), 1697, 1647 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 4.93 (s, 2H, NH2, D2O exchangeable), 6.65–6.70 (m, 1H, Ar), 7.45–7.84 (m, 4H, 3H Ar, H5, pyr.), 8.22 (d, 1H, H4, pyr.), 8.76 (d, 1H, H6, pyr.), 9.28 (s, 1H, NH, D2O exchangeable), 12.23 (s, 3H, NH, NH2, D2O exchangeable); 13C-NMR (DMSO-d6 ppm) δ: 106.48, 116.15, 121.00, 125.63, 126.15, 126.70, 127.20, 134.53, 139.21, 140.96, 148.69 (aromatic C’s), 160.32, 166.20 (2C=O); MS (EI) m/z (% rel. Int.): 272 (M+H, 52.78), 271 (M+, 57.41); Anal. Calcd for C13H13N5O2: C, 57.56; H, 4.83; N, 25.82. Found: C, 57.36; H, 4.68; N, 25.95.
2-[3-(Hydrazinecarbonyl)pyridin-2-yloxy]benzamide (3c): Yield 65%; mp 129–130°C; IR (KBr) cm−1: 3410–3201 (NH, NH2), 3124 (C–H aromatic), 1689, 1639 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 4.61 (s, 2H, NH2, D2O exchangeable), 6.45–6.50 (m, 3H, 2H Ar, H5, pyr.), 6.66–6.68 (m, 2H, 1H Ar, H4, pyr.), 8.28–8.32 (m, 2H, 1H Ar, H6, pyr.), 10.51 (s, 3H, NH, NH2, D2O exchangeable); MS (EI) m/z (% rel. Int.): 273 (M+H, 23.74), 272 (M+, 35.02); Anal. Calcd for C13H12N4O3: C, 57.35; H, 4.44; N, 20.58. Found: C, 57.24; H, 4.15; N, 20.35.
General Procedure for the Synthesis of 4a–lAn equimolar mixture of an appropriate hydrazide 3a–c and the corresponding aldehyde (1.5 mmol) each in absolute ethanol (10 mL) was heated under reflux for 4h in the presence of hydrochloric acid (2 drops). The resulting precipitate was filtered while hot, washed with water, and crystallized from ethanol to give 4a–i.
N′-(2-Bromobenzylidene)-2-(2-bromophenylamino)nicotinohydrazide (4a): Yield 80%; mp 289–290°C; IR (KBr) cm−1: 3437–3228 (NH), 1666 (C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 6.57 (t, 1H, J=7.2 Hz, Ar), 7.36–7.54 (m, 5H, Ar), 7.68–7.99 (m, 3H, 2H Ar, H5, pyr.), 8.16 (d, 1H, J=5.1 Hz, H4, pyr.), 8.44 (d, 1H, J=5.4 Hz, H6, pyr.), 8.59 (s, 1H, =CH), 12.78, 13.05 (2s, 1H, NH, D2O exchangeable, cis and trans conformers); MS (EI) m/z (% rel. Int.): 474 (M+2, 14.09); Anal. Calcd for C19H14Br2N4O: C, 48.13; H, 2.98; N, 11.82. Found: C, 48.22; H, 2.68; N, 11.98.
2-(2-Bromophenylamino)-N′-(2-chlorobenzylidene)nicotinohydrazide (4b): Yield 62%; mp 283–284°C; IR (KBr) cm−1: 3313–3201 (NH), 1670 (C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 6.56 (t, 1H, J=6.9 Hz, Ar), 7.40–7.59 (m, 5H, Ar), 7.76–8.01 (m, 3H, 2H Ar, H5, pyr.), 8.16 (dd, 1H, J=6.3, 1.8 Hz, H4, pyr.), 8.43 (dd, 1H, J=5.1, 1.8 Hz, H6, pyr.), 8.67 (s, 1H, =CH), 12.70, 13.06 (2s, 1H, NH, D2O exchangeable, cis and trans conformers); 13C-NMR (DMSO-d6 ppm) δ: 106.79, 119.46, 127.12, 127.53, 127.68, 128.16, 129.86, 130.0, 130.12, 131.42, 131.52, 133.09, 133.13, 140.32, 143.95, 145.00, 158.09, 160.31 (aromatic C’s and =CH), 162.00 (C=O); MS (EI) m/z (% rel. Int.): 429 (M+H, 2.16); Anal. Calcd for C19H14BrClN4O: C, 53.11; H, 3.28; N, 13.04. Found: C, 53.27; H, 3.43; N, 13.18.
2-(2-Bromophenylamino)-N′-(4-chlorobenzylidene)nicotinohydrazide (4c): Yield 70%; mp 185–186°C; IR (KBr) cm−1: 3444–3309 (NH), 1680 (C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 7.58 (d, 2H, J=8.4 Hz, Ar), 7.64–7.81 (m, 5H, 4H Ar, H5, pyr.), 7.97 (d, 2H, J=8.4 Hz, Ar), 8.20 (d, 1H, H4, pyr.), 8.45 (d, 1H, H6, pyr.), 8.69 (s, 1H, =CH), 10.40 (s, 1H, NH, D2O exchangeable); MS (EI) m/z (% rel. Int.): 432 (M+4, 32.60), 430 (M+2, 29.67), 428 (M+, 32.60); Anal. Calcd for C19H14BrClN4O: C, 53.11; H, 3.28; N, 13.04. Found: C, 53.42; H, 3.34; N, 13.16.
2-(2-Bromophenylamino)-N′-[4-(dimethylamino)benzylidene]nicotinohydrazide (4d): Yield 66%; mp 253–255°C; IR (KBr) cm−1: 3441–3201 (NH), 1665 (C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.98 (s, 6H, 2CH3), 6.74 (d, 2H, J=8.7 Hz, Ar), 6.90–7.55 (m, 5H, 4H Ar, H5, pyr.), 7.62 (d, 2H, J=8.7 Hz, Ar), 8.14 (d, 1H, H4, pyr.), 8.43 (d, 1H, H6, pyr.), 8.49 (s, 1H, =CH); Anal. Calcd for C21H20BrN5O: C, 57.54; H, 4.60; N, 15.98. Found: C, 57.34; H, 4.45; N, 16.08.
2-{3-[2-(2-Bromobenzylidene)hydrazinecarbonyl]pyridin-2-ylamino}benzamide (4e): Yield: 66%; mp 250–251°C; IR (KBr) cm−1: 3433–3228 (NH, NH2), 1690, 1670 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 4.05 (s, 2H, NH2, D2O exchangeable), 7.45–7.67 (m, 5H, Ar), 7.76–7.80 (m, 4H, 3H Ar, H5, pyr.), 8.16 (d, 1H, J=6.9 Hz, H4, pyr.), 8.34 (d, 1H, H6, pyr.), 8.92 (s, 1H, =CH), 11.78 (s, 1H, NH, D2O exchangeable), 11.98, 12.35 (2s, 1H, NH, D2O exchangeable, cis and trans conformers); EI-MS (% rel. abundance): 439 (M+2, 40.72), 437 (M+, 33.51); Anal. Calcd for C20H16BrN5O2: C, 54.81; H, 3.68; N, 15.98. Found: C, 54.69; H, 3.46; N, 15.78.
2-{3-[2-(2-Chlorobenzylidene)hydrazinecarbonyl]pyridin-2-ylamino}benzamide (4f): Yield: 75%; mp 260–261°C; IR (KBr) cm−1: 3441–3201 (NH, NH2), 1693, 1670 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 4.05 (s, 2H, NH2, D2O exchangeable), 6.68 (t, 1H, J=7.2 Hz, Ar), 7.35–7.93 (m, 8H, 7H Ar, H5, pyr.), 8.14 (d, 1H, J=6.3 Hz, H4, pyr.), 8.23 (d, 1H, H6, pyr.), 8.58 (s, 1H, =CH), 11.81 (s, 1H, D2O exchangeable), 12.37, 13.31 (2s, 1H, NH, D2O exchangeable, cis and trans conformers); EI-MS (% rel. abundance): 393 (M+, 1.50); Anal. Calcd for C20H16ClN5O2: C, 60.99; H, 4.09; N, 17.78. Found: C, 61.23; H, 4.18; N, 17.95.
2-{3-[2-(4-Chlorobenzylidene)hydrazinecarbonyl]pyridin-2-ylamino]benzamide (4g): Yield: 60%; mp 258–259°C; IR (KBr) cm−1: 3440–3250 (NH, NH2), 1700, 1662 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 4.03 (s, 2H, NH2, D2O exchangeable), 6.68 (t, 1H, J=7.8 Hz, Ar), 7.48–7.86 (m, 8H, 7H Ar, H5, pyr.), 8.14 (d, 1H, J=6.3 Hz, H4, pyr.), 8.23 (d, 1H, J=5.4 Hz, H6, pyr.), 8.75 (s, 1H, =CH), 11.69 (s, 1H, NH, D2O exchangeable), 12.36, 13.32 (2s, 1H, NH, D2O exchangeable, cis and trans conformers); EI-MS (% rel. abundance): 397 (M+4, 40.84), 395 (M+2, 43.98), 394 (M+H, 28.80); Anal. Calcd for C20H16ClN5O2: C, 60.99; H, 4.09; N, 17.78. Found: C, 60.87; H, 4.27; N, 17.98.
2-{3-[2-(4-Dimethylaminobenzylidene)hydrazinecarbonyl]pyridin-2-ylamino}benzamide (4h): Yield: 76%; mp 254–255°C; IR (KBr) cm−1: 3302–3217 (NH, NH2), 1697, 1667 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.96 (s, 6H, 2CH3), 3.98 (s, 2H, NH2, D2O exchangeable), 6.68 (t, 1H, J=7.8 Hz, Ar), 7.48–7.86 (m, 8H, 7H, Ar, H5, pyr.), 8.14 (d, 1H, J=6.3 Hz, H4, pyr.), 8.21 (d, 1H, J=5.7 Hz, H6, pyr.), 8.75 (s, 1H, =CH), 11.33 (s, 1H, NH, D2O exchangeable), 12.38, 13.32 (2s, 1H, NH, D2O exchangeable, cis and trans conformers); 13C-NMR (DMSO-d6 ppm) δ: 40.34 (2CH3), 106.47, 116.15, 121.00, 121.10, 125.84, 126.15, 126.20, 127.15, 127.20, 134.53, 139.21, 140.96, 148.81, 150.16, 159.25, 159.34 (aromatic C’s and =CH), 160.22, 160.31 (2C=O); EI-MS (% rel. abundance): 403 (M+H, 15.07), 402 (M+, 13.88); Anal. Calcd for C22H22N6O2: C, 65.66; H, 5.51; N, 20.88. Found: C, 65.48; H, 5.80; N, 20.65.
2-{3-[2-(2-Bromobenzylidene)hydrazinecarbonyl]pyridin-2-yloxy}benzamide (4i): Yield 70%; mp 230–231°C; IR (KBr) cm−1: 3435–3220 (NH, NH2), 1700, 1666 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 6.56 (t, 1H, J=6.9 Hz, Ar), 7.34–7.50 (m, 5H, Ar), 7.68–7.70 (m, 3H, 2H Ar, H5, pyr.), 7.81 (br s, 2H, NH2, D2O exchangeable), 7.99 (dd, 1H, J=5.7, 2.1 Hz, H4, pyr.), 8.44 (dd, 1H, J=5.4, 1.8 Hz, H6, pyr.), 8.61 (s, 1H, =CH), 12.72, 13.07 (2s, 1H, NH, D2O exchangeable, cis and trans conformers); MS (EI) m/z (% rel. Int.): 438 (M+, 1.51); Anal. Calcd for C20H15BrN4O3: C, 54.69; H, 3.44; N, 12.75. Found: C, 54.55; H, 3.57; N, 12.68.
2-{3-[2-(2-Chlorobenzylidene)hydrazinecarbonyl]pyridin-2-yloxy}benzamide (4j): Yield 75%; mp 276–277°C; IR (KBr) cm−1: 3420–3194 (NH, NH2), 1678, 1670 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 6.57 (t, 1H, J=6.9 Hz, Ar), 7.43–7.55 (m, 5H, Ar), 7.75–7.78 (m, 3H, 2H Ar, H5, pyr.), 7.81 (s, 2H, NH2, D2O exchangeable), 8.01 (dd, 1H, J=6.9, 2.4 Hz, H4, pyr.), 8.44 (dd, 1H, J=5.1, 2.4 Hz, H6, pyr.), 8.65 (s, 1H, =CH), 12.76, 13.09 (2s, 1H, NH, D2O exchangeable, cis and trans conformers); Anal. Calcd for C20H15ClN4O3: C, 60.84; H, 3.83; N, 14.19. Found: C, 60.77; H, 3.52; N, 14.45.
2-{3-[2-(4-Chlorobenzylidene)hydrazinecarbonyl]pyridin-2-yloxy}benzamide (4k): Yield 68%; mp 181–182°C; IR (KBr) cm−1: 3429–3360 (NH, NH2), 1674, 1651 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 7.56 (d, 2H, J=8.7 Hz, Ar), 7.66–7.72 (m, 5H, 4H Ar, H5, pyr.), 7.88 (d, 2H, J=8.7 Hz, Ar), 8.22 (d, 1H, H4, pyr.), 8.50 (d, 1H, H6, pyr.), 8.71 (s, 1H, =CH); MS (EI) m/z (% rel. int.): 396 (M+2, 23.37), 394 (M+, 19.29); Anal. Calcd for C20H15ClN4O3: C, 60.84; H, 3.83; N, 14.19. Found: C, 60.79; H, 4.00; N, 14.32.
2-{3-[2-(4-Dimethylamino)benzylidene)hydrazinecarbonyl)pyridin-2-yloxy)benzamide (4l): Yield 70%; mp 264–265°C; IR (KBr) cm−1: 3440–3325 (NH, NH2), 1674, 1651 (2C=O); 1H-NMR (DMSO-d6, 300 MHz) δ (ppm): 2.97 (s, 6H, 2CH3), 6.54 (t, 1H, J=7.2 Hz, Ar), 6.72–6.76 (m, 4H, Ar), 7.55–7.59 (m, 4H, 3H Ar, H5, pyr.), 7.77 (d, 1H, J=6.0 Hz, H4, pyr.), 8.19 (s, 1H, =CH), 8.43 (d, 1H, J=5.1 Hz, H6, pyr.), 12.64, 12.76 (2s, 1H, NH, D2O exchangeable, cis and trans conformers); 13C-NMR (DMSO-d6 ppm) δ: 40.33 (2CH3), 106.70, 111.60, 111.69, 119.85, 119.90, 121.38, 121.44, 128.57, 139.60, 144.28, 144.36, 148.96, 151.35, 151.55, 159.35, 159.40 (aromatic C’s and =CH), 162.08, 162.12 (2C=O); MS (EI) m/z (% rel. Int.): 401 (M-2, 1.51); Anal. Calcd for C22H21N5O3: C, 65.50; H, 5.25; N, 17.36. Found: C, 65.78; H, 5.33; N, 17.18.
Pharmacological Activity. AnimalsThe investigations were carried out using healthy male Swiss albino mice with a weight of 20–25 g. Mice were supplied by the Modern Veterinary Office for Laboratory Animals (Cairo, Egypt). All animals were naive to drug treatment and experimentation at the beginning of the study. Mice were allowed to acclimatize for 1–2 weeks prior to use, with standard pellet diet and water ad libitum. The animals were randomly assigned to experimental groups, 6 mice each. All experiments were conducted between 10:00 a.m. and 16:00 p.m. to eliminate circadian influence on animal behaviour. All experimental protocols were approved by the Institutional Animal Care and Use Committee at the Faculty of Pharmacy, Suez Canal University.
Preparation of the Tested Compounds and the Standard DrugTest compounds were given orally to test animals after suspending in a 1% sodium carboxymethylcellulose (CMC) aqueous solution. The control group animals received the same experimental handling as those of the test groups except that the drug treatment was replaced with appropriate volumes of the dosing vehicle. Mefenamic acid (25 mg/kg) was kindly provided by Al-Qahira Pharmaceutical Company (Cairo, Egypt). It was prepared in 1% CMC and used as a reference drug.
Analgesic ActivityAnalgesic activity was carried out by acetic acid induced writhing method.21) Aqueous acetic acid solution (0.1 mL, 1%, i.p.) was used to induce writhing. Mice were kept individually in the test cage, before acetic acid injection and habituated for 30 min. Screening of analgesic activity was performed after p.o. administration of test drugs at the dose of 25 mg/kg. All compounds were dissolved in 1% CMC solution. One group was kept for the control experiment and received p.o. administration of 1% CMC. Mefenamic acid was used as a reference drug. After 30 min of drug administration, acetic acid solution (0.1 mL, 1%, i.p.) was given to each mouse.
Stretching movements consisting of arching of the back, elongation of body and extension of hind limbs were counted for 5–15 min after acetic acid injection. The analgesic activity was expressed as follows:
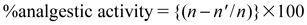 | (1) |
where n=mean number of writhes of control group and n′=mean number of writhes of test group.
Anti-inflammatory ActivityAnti-inflammatory activity test was performed following the method of Winter et al.22) Carrageenan (Sigma, St. Louis, Missouri, U.S.A.) was freshly prepared as suspension (0.05 mL, 1% w/v solution in 0.9% saline). Carrageenan solution was injected into the subplantar tissue of the right hind paw of each mouse. Control animals were injected with saline into that of the left hind paw.
Animals were divided in groups of six each. One group was kept as control and the animals of other groups were pre-treated with the test drugs suspended in 1% CMC and given orally 30 min prior carrageenan injection. The paw volume was measured before and after 4 h of carrageenan injection. The difference in thickness between the right and left hind paw was measured with a pair of dial thickness gauge callipers (Ozaki Co., Tokyo, Japan). Mean values of treated groups were compared to those of control group and analysed using statistical methods. The percentage inhibition of inflammation was calculated by applying the following formula:
 | (2) |
where Vc=edema volume in control group, Vt=edema volume in groups treated with the test compounds.
Furthermore, the mice were anesthetized with a mixture of ketamine (50 mg/kg, i.p.)/xylazine (10 mg/kg, i.p.), then sacrificed by decapitation. Thereafter, a midline incision was made, and blood samples were withdrawn from the heart. Thirty minutes after collection, blood samples were processed by centrifugation at 2000×g for 15 min. Serum samples were separated, collected in clean tubes and stored at −80°C until being used for enzyme-linked immunosorbent assay (ELISA) assays. ELISA kits for IL-6 (Glory Science Co., Ltd., Del Rio, TX, U.S.A.) was used for determination of tissue levels of this marker. The assays were carried out following the instructions of the manufacturer using an automated ELISA reader (Europe S.A., Belgium).
Acute UlcerogenesisAcute ulcerogenesis test was determined according to Cioli et al.24) Ulcerogenic activity was evaluated after p.o. administration of the test compound or mefenamic acid at the dose of 50 mg/kg. Control mice were treated orally with the vehicle (suspension of 1% CMC). Food but not water was removed 24 h before administration of the test compounds. After the drug treatment, mice were fed normal diet for 17 h. Mice were anesthetized with a mixture of ketamine (50 mg/kg, i.p.)/xylazine (10 mg/kg, i.p.), then sacrificed by decapitation. The stomach was removed and opened along the greater curvature, washed with distilled water and cleaned gently by dipping in saline. The gastric mucosa of the mice was examined by means of a magnifying glass. For each stomach, the severity of mucosal damage was assessed by measuring severity index i.e., severity of drug to cause mucosal damage, which is the difference between the mean score of each treated group and the mean score of the control group.
Immunohistochemistry25)Three different sections were cut on positively charged slides and fixed in a 65°C oven for 1 h. The slides were placed in a coplin jar filled with 60 mL of triology (Cell Marque®, CA, U.S.A.) working solution and the jar is securely positioned in an autoclave. The autoclave was kept at a temperature of 120°C for 15 min. The pressure was released, the coplin jar was removed and left to cool for 30 min. Sections were washed and immersed in Tris-buffered saline to adjust the pH. This was repeated between each step of the immunohistochemical procedure. Quenching endogenous peroxidase activity was performed by immersing slides in 3% hydrogen peroxide for 10 min.
Excess serum was drained from each slide without washing, and 2–3 drops of the primary antibodies against myeloperoxidase (Thermo Fischer Scientific®, Fremont, CA, U.S.A.) were added. The slides were incubated in the humidity chamber for 1 h. Henceforward, biotinylated secondary antibody was applied to each slide for 20 min, followed by another 20 min incubation with the enzyme conjugate. Three drops of 3,3′-diaminobenzidine (DAB) chromogen were added to each slide for 2 min. DAB was rinsed and the slides were counterstaining with Mayer’s hematoxylin. Cover slipping was performed as the final steps before slides being examined using a light microscope (Olympus CX21, Japan). The count of the optical density was carried out by means of a computer assessed image analysis system “Image J 1.45F” (National Institute of Health, U.S.A.) on five randomly selected regularly spaced sections (4×). The average optical density in each slide was used to calculate the relative optical density compared to the vehicle group.
Statistical AnalysisData was collected, tabulated and expressed as mean±S.E.M. Statistical differences between the treatments and the control were evaluated using one-way ANOVA followed by Tukey’s test for multiple comparisons. All statistical analyses were carried out using The Statistical Package for Social Sciences, version 17 (SPSS Inc., Chicago, IL, U.S.A.). p<0.05 was considered to be statistically significant.
Authors wish to acknowledge Al-Qahira Pharmaceutical Company for the generous gift of mefenamic acid.