Abstract
Certain antimicrotubule agents displaying colchicine-like tubulin-depolymerizing activity can act as both cytotoxic and vascular-disrupting agents (VDAs). VDAs constitute a new class of anticancer drugs and are currently in clinical trials. We have developed a VDA clinical candidate (phase II) diketopiperazine (DKP)-type antimicrotubule agent called plinabulin (7) derived from the natural DKP phenylahistin (5), which displays colchicine-like tubulin-depolymerizing activity. To develop more potent antimicrotubule DKP derivatives, we performed an intensive structure–activity relationship study examining the phenyl group of compound 7. This study identified more potent DKP derivatives (2,5-difluoro derivatives [29] and benzophenone derivatives [36]) with vascular-disrupting activities. The benzophenone moiety of compound 36 was further modified to yield the most potent cytotoxic derivative yet discovered, the 4-fluorobenzophenone derivative 38m, which inhibited the growth of HT-29 cells in vitro at subnanomolar levels. As both VDAs and cytotoxic agents, these potent DKP derivatives are valuable second-generation drug candidates. The chemical biology of plinabulin was examined by designing and synthesizing biotin-tagged photoaffinity probes 40–42 that could be used to indicate the binding mode of compound 7 with tubulin. A tubulin photoaffinity labeling study suggested that compound 7 binds at the interface between the α- and β-tubulins near the colchicine-binding site and not inside the colchicine-binding cavity. A water-soluble prodrug of the poorly water-soluble 7 was next designed in an effort to improve the pharmacokinetics and chemotherapeutic indices. The lead compound 56 revealed high water solubility and a half-life profile appropriate for an injected drug.
1. Introduction
Microtubules, which are noncovalently linked polymers formed by the α- and β-tubulin heterodimers, are a major component of the cytoskeleton with an important role in a variety of cellular functions, such as cell shape maintenance, intracellular transport, polarity, cell signaling, and mitosis.1) The crucial functions of the cytoskeletal system make microtubules attractive targets for anticancer chemotherapy. To date, numerous antimicrotubule agents with antimitotic properties and anticancer potential have been discovered and developed. Most of these antimicrotubule agents are classified into three major groups.2) The first group contains the microtubule-stabilizing taxanes (e.g., paclitaxel and dosetaxel), which recognize the taxol domain on tubulin. The second and third groups are the microtubule-depolymerizing vinca alkaloids (e.g., vinblastine, vincristine) and colchicine analogues (e.g., colchicine [1, Fig. 1], combretastatins, and podophillotoxin), which recognize the vinca and colchicine domains on tubulin, respectively. Taxanes and vinca alkaloids from the first and second groups have been used clinically for cancer chemotherapy,3) whereas the colchicine analogues have not yet been approved for clinical use due to their extreme toxicity, except for an antigout drug.4) In the 1990s, several agents possessing colchicine-like tubulin-depolymerizing activity, such as the combretastatin A-4 phosphate prodrug (CA4P, 3, Fig. 1),5,6) AVE8062,7) and ABT-751,8) were recognized as having the characteristics of both cytotoxic and vascular-disrupting agents (VDAs).9) VDAs form a new class of anticancer drugs that induce the collapse of established tumor vasculature via rapid microtubule depolymerization. This action leads to a loss of blood supply and eventual contraction of the tumor. More than 10 antimicrotubule agents with vascular-disrupting activity are currently progressing to clinical trials for the treatment of cancer.
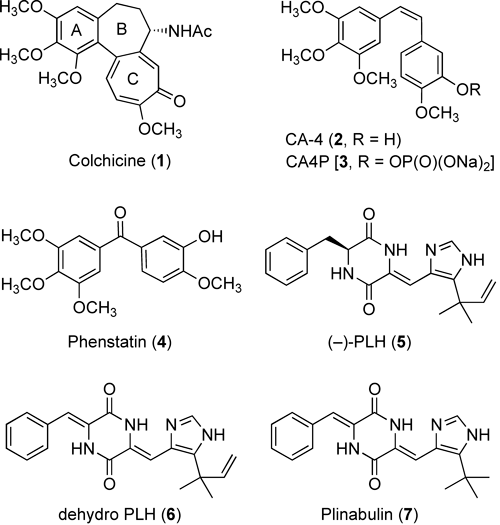
Fig. 1. Chemical Structures of Antimicrotubule Agents 1–7
During the development of our original VDA candidate, we focused on a low molecular-weight natural diketopiperazine (DKP) product, (−)-phenylahistin [(−)-PLH, 5, Fig. 1], which was isolated from Aspergillus ustus by Kanoh et al. in 1997 and specifically inhibits the cell cycle in the G2/M phase by inhibiting tubulin polymerization.10,11) A competitive binding assay using [3H]colchicine indicated that compound 5 recognized the colchicine-binding site on tubulin.12) After structurally modifying compound 5, we developed a DKP-type VDA, NPI-2358 (7, Fig. 1) in 2006.13) Compound 7 was chosen as a clinical candidate with the United States adopted names designation plinabulin.14,15) In this article, we review our recent achievements in the medicinal chemistry and chemical biology of the novel DKP-type antimicrotubule agent 7, including the results of extensive structure–activity relationship (SAR) studies, the synthesis of its chemical probes, a tubulin photoaffinity-labeling study to understand the binding mode of the antimicrotubule DKP, and the development of water-soluble prodrugs using click chemistry to enhance the practical significance of the clinical candidate plinabulin (7).
2. Development of Plinabulin (7) from the Natural DKP (−)-Phenylahistin (5)
In a previous study of the chemically modified or totally synthesized compound 5 and its derivative,16,17) the structural requirements, which are important for eliciting potent cytotoxicity, were understood as including: 1) a rigid and planar pseudo-tricyclic structure formed by a hydrogen bond between the DKP and imidazole rings; 2) the L-form of the Phe residue; and 3) the gem-dimethyl structure at the 5-position of the imidazole ring (Fig. 2). The olefin structure at the dimethylallyl group was not found to be important, and alkylation of the τ-nitrogen of the imidazole ring could be tolerated with respect to the activity.

Fig. 2. Structural Entities Required for the Potent Biological Activity of (−)-PLH (5)
Kanzaki et al. reported that a biotransformed derivative of compound 1 with an additional α,β-unsaturated unit joining the DKP core with the benzyl substituent (dehydrophenylahistin, ΔPLH, 6, Fig. 1) showed a significant increase in the antipronuclear fusion activity in sea urchin eggs.18) This information was used to develop the tert-butyl derivative (7, KPU-2/NPI-2358, Fig. 1).13) Compound 7 showed potent cytotoxicity (IC50 value=15 nM) against HT-29 cell lines, similar to the activity of colchicine. The potent antimicrotubule activities of compound 7 were observed in a microtubule polymerization assay, and cytotoxic activity was observed against HeLa cells. Compound 7 exhibited biological effects similar to those of other antimicrotubule agents (e.g., paclitaxel and nocodazole), as the cell cycle progression of HeLa cells was inhibited in the G2/M phase. Furthermore, compound 7 dose dependently increased human umbilical vein endothelial cell (HuVEC) monolayer permeability in an in vitro model of tumor vascular collapse.13) These results supported the selection of the novel DKP-type VDA 7 as a clinical candidate.
3. SAR Study of Plinabulin
3.1. Synthetic Route of Plinabulin and Its Derivatives3.2. Modification of the Alkyl Group on the Imidazole RingThe effects of the alkyl substitution on the 5-position of the imidazole ring are summarized in Table 1. The derivatives with normal alkyl groups, i.e., methyl (compound 17), n-propyl (compound 18), and n-butyl (compound 19) groups, showed significantly lower potencies than compound 7; however, compound 20, which included a branched alkyl group (the isopropyl group), exhibited comparable but slightly weaker potency compared with compound 7, which included a tert-butyl group at the same position. These results suggested that the branched chain at the 5-position of the imidazole ring was critical to the potent activity. Therefore, we adopted the tert-butyl group in subsequent derivative designs.
Table 1. Cytotoxic Activities of the Plinabulin Derivatives with Substitutions at the 5-Position of the Imidazole Ring
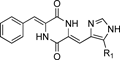 |
|---|
| Compound | R1 | IC50 (nM)/HT-29a) |
|---|
| 1 [COL] | | 16±3.0 |
| 7 | tert-Butyl | 14.9±3.8 |
| 17 | Methyl | 339±41 |
| 18 | n-Propyl | 153±18 |
| 19 | n-Butyl | 112±16 |
| 20 | Isopropyl | 15.5±1.9 |
a) Values indicate the mean±S.E.M. from at least three independent dose–response curves.
The effects of phenyl ring substitutions were next examined (Table 2). A single substitution with a methoxy group at the o-position (compound 21) resulted in a 5-fold decrease in cytotoxicity, whereas m-substitution (compound 22) maintained a potency comparable with that of compound 7. The p-substitution (compound 23) resulted in a marked decrease in potency (>400-fold). These results indicated that the meta-position may potentially be used to modify the potency. A double substitution of methoxy groups (compounds 24, 25) on the phenyl ring resulted in a marked decrease in potency. The triple methoxy derivatives, similar to those occurring in colchicine, also demonstrated very low potencies (data not shown).
Table 2. Cytotoxic Activities of the Plinabulin Derivatives with Substitutions on the Phenyl Ring
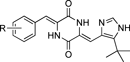 |
|---|
| Compound | R | IC50 (nM)/HT-29a) |
|---|
| 7 | H | 14.9±3.8 |
| 21 | 2-OMe | 75.5±2.5 |
| 22 | 3-OMe | 26.1±8.5 |
| 23 | 4-OMe | 6570 |
| 24 | 2,3-diOMe | 868 |
| 25 | 3,4-diOMe | 2020 |
| 26 | 2-F | 30.0±17 |
| 27 | 3-F | 13.1±5.3 |
| 28 | 4-F | 501 |
| 29 | 2,5-diF | 2.6±0.5 |
| 30 | 3-Cl | 24.5±8.7 |
| 31 | 3-Br | 31.1±7.3 |
| 32 | 3-NO2 | 44.7±11.9 |
| 33 | 3-CF3 | 44.0±7.49 |
| 34 | 3-OH | 364 |
| 35 | 3-Vinyl | 17.0±1.5 |
| 36 | 3-Benzoyl | 1.4±0.4 |
a) Values indicate the mean or mean±S.E.M. (for potent compounds) from at least three independent dose–response curves.
An investigation of the fluoro substitutions revealed that the 3-fluoro derivative (27) exhibited potent cytotoxic activity, and the 2,5-difluoro derivative (29) showed a 5.7-fold increase in potency (IC50 value=2.6 nM) compared with compound 7. This may reflect the unique properties of fluorinated organic compounds, including the improved entropies associated with the ligand binding to the target.26)
The methoxy and fluoro substitution results suggested that modifications at the meta-positions on the phenyl ring could increase the potency; therefore, a variety of substituent groups (Cl, Br, OH, NO2, CF3, vinyl, benzoyl, etc.) were introduced to the meta-position to test their effects on potency. The 3-nitro derivative (32) and the 3-trifluoro derivative (33) slightly decreased the potency. The 3-hydroxyl derivative (34) significantly reduced the potency. By contrast, the 3-vinyl derivative (35) displayed potent activity (IC50=17 nM), equipotent to compound 7. The 3-benzoyl derivative (36) in particular provided markedly potent activity with 11-fold more potent cytotoxicity (IC50 value=1.4 nM) than was observed for compound 7. The enhanced potency of the benzophenone derivative 36 could reflect, in part, an entropic effect with improved binding energy provided by conformational restrictions to an active form most suitable for binding. The benzoyl moiety may also have increased the interaction surface to stabilize binding further. The hydrophobic properties of the benzophenone moiety in compound 36 may have increased the cell penetration efficiency and bolstered the highly potent cytotoxic activity (Table 2), with almost no enhancement of the tubulin-binding potency, as shown in Table 4.
3.4. Modification of the Benzophenone MoietyEncouraged by the results obtained from replacing the phenyl ring with a benzophenone moiety, we further modified the benzophenone moiety of compound 36. Several benzophenone-type CA-4 analogues, such as phenstatin27) (see Fig. 1), have been reported to display relatively potent antimicrotubule activities, and their activities increase upon introducing substitutions into the benzophenone moiety.28–30) Further modifications and structural optimization at the benzophenone of compound 36 were thought to be valuable for developing more potent derivatives and for understanding the pharmacophore in the DKP-type VDA.
As shown in Table 3, in the case of a single methoxy substitution, the 2-substituted derivative (38f) and the 3-substituted derivative (38g) resulted in a marked decrease in cytotoxic activity (257-, 28-fold, respectively); however, the 4-substituted derivative (38h) essentially retained the original potency. The single modification with a methoxy group at the 4-position was generally well tolerated, suggesting an opportunity for further modification. Double substitution with methoxy groups at the 2,4- or 3,4-positions (38i, j) led to a dramatic decrease in potency. Triple substitutions, such as a 3,4,5-methoxy substitution, did not show significant potencies (data not shown).
Table 3. Cytotoxic Activities of the Benzophenone Derivatives of Plinabulin with a Substitution on the Phenyl Ring
 |
|---|
| Compound | R | IC50 (nM)/HT-29a) |
|---|
| 36 | H | 1.4±0.4 |
| 38f | 2-OMe | 360±66 |
| 38g | 3-OMe | 39±12 |
| 38h | 4-OMe | 3.8±0.4 |
| 38i | 2,4-diOMe | 1800 |
| 38j | 3,4-diOMe | 7300 |
| 38k | 2-F | 3.0±2.0 |
| 38l | 3-F | 0.6±0.1 |
| 38m | 4-F | 0.5±0.1 |
| 38n | 4-Cl | 1.1±0.0 |
| 38o | 4-Br | 4.0±0.7 |
| 38p | 4-CF3 | 147±15 |
a) Values indicate the mean or mean±S.E.M. (for potent compounds) determined from at least three independent dose–response curves.
An examination of the effects of a single fluorine atom substitution revealed that the 3- and 4-substituted derivatives (38l, m) showed 2.3- or 2.8-fold increases in potency compared with compound 36, respectively; however, the 2-substituted derivatives (38k) displayed a slight reduction in potency. Modification at the 4-position generally resulted in greater potency; therefore, we further examined the effects of replacements at this position. The substituted derivatives bearing chloro, bromo, or trifluoromethyl groups at the 4-position (38n–p) did not display potency superior to that of compound 38m. These results indicated that a modification with a fluorine atom at the 4-position was acceptable for increasing the potency; however, modifications with bulky electron-withdrawing groups, such as Br and CF3, were not appropriate. These results also suggested that tubulin could recognize the benzophenone moiety of compound 36 rigorously, and the space available to accommodate a substituent was extremely limited.
General antimicrotubule agents, such as colchicine, podophyllotoxin, combretastatin, and phenstatin analogues, which recognize the same binding site on β-tubulin as is recognized by (−)-PLH (5), are structurally characterized as having multiple methoxy groups that make marked contributions to their potent antimicrotubule activities31–34); however, multiple methoxy substitutions of a plinabulin derivative (e.g., compounds 24, 25, 38i, j) displayed low potencies. These results suggested that plinabulin and its derivatives recognized tubulin in a manner that differed from the binding by colchicine and its biological homologues.
3.5. Inhibition of Microtubule Polymerization and Tubulin-Binding AbilityThe 2,5-difluoro derivative (29), benzophenone derivative (36), and 4-fluorobenzophenone derivative (38m) showed higher cytotoxic potencies against HT-29 cells than compound 7; therefore, we tested these compounds in a fluorescence-based tubulin-binding assay35) and microtubule depolymerization assay using purified porcine tubulin (Table 4). In the tubulin depolymerization assay, compounds 29, 36, and 38m exhibited more potent tubulin-binding affinities and microtubule-depolymerization activities than compound 7. The tubulin-depolymerizing abilities were ordered 29>36≥38m, which was not consistent with their cytotoxic activities against HT-29 cells. The introduction of a fluorine atom (38m) to compound 36 did not increase the microtubule-depolymerizing abilities, although the cytotoxicity against HT-29 cells was 3-fold more potent than the cytotoxicity of compound 36. The observed increase in the cytotoxicity of compound 38m at the cellular level may have been due to an improvement in the cell permeability caused by the introduction of the hydrophobic fluorine atom. Although the tubulin-binding constants, Kd, for compounds 29 and 36 were lower than the value of compound 7, compound 36, with a benzophenone structure, bound relatively weakly to tubulin given the measured cytotoxicity of the compound. The potency of compound 36 with respect to inducing cell cycle arrest in the G2/M phase, as characterized by flow cytometry, exceeded the potency of compound 29. These results suggest that the benzophenone derivative functioned through a distinct mode of action to induce such potent cytotoxicity. For example, this derivative may display better cell penetration, in addition to displaying tubulin-depolymerization activity.
Table 4. Inhibition of
in Vitro Tubulin Polymerization and Tubulin Binding Activity
| Compound | Inhibition of tubulin polymerization IC50 (µM)a) | Tubulin binding Kd (µM)a) |
|---|
| 1 [COL] | 4.12±0.63 | 3.3±0.3 |
| 2 [CA-4] | 0.55±0.01 | N.D.b) |
| 7 | 1.31±0.10 | 1.0±0.5 |
| 29 | 0.51±0.02 | 0.20±0.11 |
| 36 | 0.76±0.14 | 0.62±0.07 |
| 38m | 0.90±0.03 | N.D.b) |
a) Values indicate the mean±S.E.M. determined from at least three independent dose–response curves. b) N.D., not determined.
The vascular-disrupting functions of the plinabulin derivatives were examined by measuring the cytotoxicities and antimicrotubule activities of compounds 29 and 36 against HuVECs in an in vitro model of tumor vascular endothelial cells13) (Table 5). Compounds 29 and 36 exhibited more potent activities than compounds 2 and 7 in terms of both cytotoxicity and microtubule depolymerization against HuVECs, suggesting that these derivatives were more effective VDAs than the existing compounds. Compounds 29 and 36 are valuable second-generation drug candidates, displaying both vascular-disrupting and cytotoxic activities.
Table 5. Antimicrotubule and Cytotoxic Activities against HuVECs
| Compound | IC50 (nM)/HuVECsa) | 30-min HuVEC microtubule depolymerization |
|---|
| ED100 (nM) b) | Dose tested (nM) |
|---|
| 2 [CA-4] | 2.4±0.8 | 10 | 2, 10, 20, 50 |
| 7 | 11±0.6 | 3 | 3, 5, 10 |
| 29 | 1.5±0.3 | 2 | 1, 2, 5, 50 |
| 36 | 0.5±0.1 | 1 | 0.1, 1, 5, 10 |
a) Values indicate the mean±S.E.M. determined from at least three independent dose–response curves. b) Lowest concentration tested at which complete depolymerization was observed.
4. Chemical Biology Approach to Understand the Mode of Action of Plinabulin
(−)-PLH (5), a lead derivative of plinabulin (7), competitively inhibits the binding of colchicine to tubulin, as observed in a radiolabeled binding assay using [3H]colchicine12); however, the three-dimensional structures of colchicine (1) and compound 7 failed to superimpose favorably in a computational molecular modeling study.36) The conformation of 7 tended to be more extended than that of 5 due to a structural conversion from the monodehydroDKP to the didehydroDKP. The biological activities of a series of compound 7 derivatives in which a methoxy substitution was introduced onto the phenyl ring, in combination with the biological activities of compound 36 derivatives in which a methoxy substitution was introduced onto the benzophenone moiety as described in Section 3, suggested that the mode by which compound 7 recognizes tubulin differed from the mode by which colchicine and its biological homologues recognize tubulin. The precise binding mode of compound 7 and the mechanism by which it depolymerizes microtubules have not yet been elucidated; therefore, we investigated the binding mode at the molecular level using a chemical biology approach in an effort to understand the mechanism by which DKP-type antimicrotubule agents depolymerize microtubules.36–38) These studies can potentially accelerate the drug development process by characterizing the features that are important for molecular recognition.
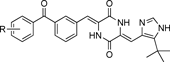
4.1. Design and Synthesis of Chemical ProbesThe SAR study of (−)-phenylahistin (5) and plinabulin (7) was designed with the understanding that the τ−nitrogen of the imidazole ring was not important for biological activity. The SAR study identified a potent oxazole derivative 39 (IC50 value=3.9 nM against HT-29 cells) with a benzophenone structure (Fig. 3). A biotin-tagged chemical probe based on the structure of compound 39 was synthesized given that: 1) the oxazole synthon 10a can be easily obtained as an intermediate in the synthesis of compound 7 (Chart 1); 2) the oxazole oxygen is generally not a reaction center in the presence of the imidazole nitrogen; and 3) the benzophenone structure is widely used for protein photoaffinity labeling.39–43) The biotin tag could be readily detected using an avidin–peroxidase system based on the avidin–biotin binding interaction.44,45) The tubulin photoaffinity labeling study was performed by synthesizing three types of chemical probe. The first chemical probe comprised compound 40 and a biotin tag at the 4-position of the benzophenone ring in 39, and the second chemical probe comprised compound 41 with a linker46) that was longer than the linker present in 40. The third chemical probe comprised compound 42 with a biotin tag at the 5-position of the oxazole ring (Fig. 4).

Fig. 3. Structure of Photoreactive Benzophenone Derivative 39
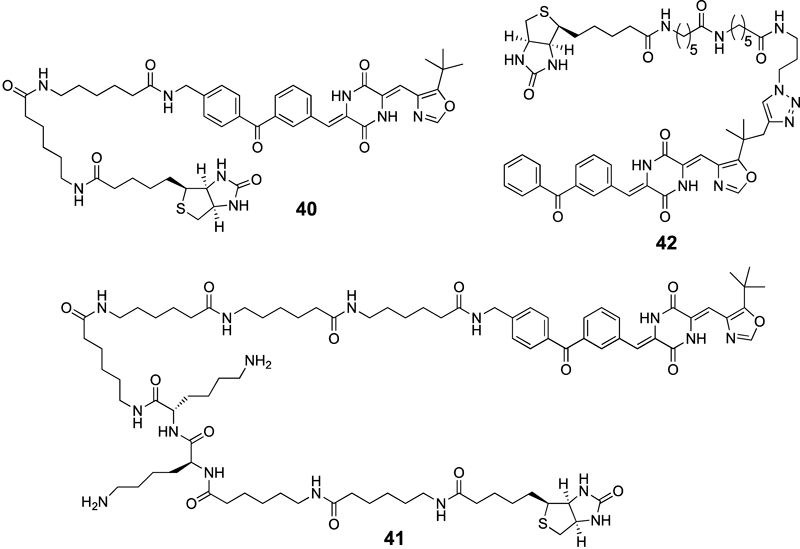
Fig. 4. Structures of Chemical Probes 40–42
The biological activities of probes 40–42 were evaluated using a fluorescence-based purified tubulin-binding assay35) or a cytotoxicity assay against HT-29 cells. The binding assay was performed using bovine or porcine purified tubulin in 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 6.8) at 37°C, and the dissociation constants (Kd) were determined based on the change in the intrinsic fluorescence derived from the tryptophan residues of the tubulin. The Kd values of probes 40–42 were calculated to be 7.2, 25.2, and 17.9 µM, respectively (Table 6). Although biologically significant, the binding affinities of probes 40–42 were about 7-, 25-, or 18-fold weaker than the binding affinity of compound 7 (1.06 µM), respectively. We next examined the cytotoxicity of probes 40 and 42 using an in vitro cytotoxicity assay against HT-29 cells. In this assay, probes 40 and 42 showed a low but significant cytotoxicity against the HT-29 human colon cancer cell line with IC50 values of 354 nM and 9.8 µM, respectively (Table 6). The higher molecular weight and greater hydrophilicity of probes 40 and 42 probably reduced the rate of cellular uptake, thereby reducing the cytotoxicity.
Table 6. Biological Activities of Probes
40–
42| Compound | Kd (µM)a) | Cytotoxicity IC50 (nM)a) |
|---|
| 40 | 7.2±1.0 | 354±29 |
| 41 | 25.2±8.2 | ntb) |
| 42 | 17.9±3.4 | 9788±1680 |
| 7 [Plinabulin] | 1.0±0.5 | 15±4 |
a) Values indicate the mean±S.E.M. determined from at least three independent dose–response curves. b) nt, not tested.
A pull-down assay was performed using probe 41-conjugated streptavidin beads from the human fibrosarcoma (HT-1080) cell lysate in order to understand the binding affinity of probe 41 toward human tubulin. The HT-1080 whole-cell lysate was incubated with photoaffinity probe 41-conjugated beads at 4°C overnight, and the proteins retained on the beads were analyzed using Western blotting. Figure 5 shows that the α- and β-tubulins were found to bind to probe 41 (lane 2), although they did not appear to bind to the biotinylated 41 conjugated to the beads or to the unconjugated beads as negative controls (lanes 3 and 4). Binding between probe 41 and β-actin, another major protein component of the cytoskeleton in eukaryotic cells,47) was not observed (lower panel in Fig. 5). These results suggested that probe 41 selectively binds to human tubulin in the whole-cell lysate.
4.3. Tubulin Photoaffinity LabelingA tubulin photoaffinity-labeling study was performed once chemical probes 40–42 were found to bind to tubulin. Purified bovine or porcine tubulin was photoirradiated at 365 nm in the presence or absence of probes 40–42 on ice after incubation at 37°C in MES buffer (containing 1 mM guanosine 5′-triphosphate (GTP), pH 6.8) for the appropriate period of time. The samples were then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), the proteins were transferred to a nitrocellulose or a polyvinylidene difluoride (PVDF) membrane, and the photolabeled proteins were visualized using a streptavidin-horseradish peroxidase (HRP)/enhanced chemiluminescence (ECL) system. The photoirradiated samples displayed UV irradiation time-dependent labeling by probes 40–42 (Figs. 6A, 7A), although low levels of nonspecific photolabeling by the benzophenone were also observed. Interestingly, probe 41-labeled tubulin displayed two clearly separated bands, whereas probe 40, which included a shorter linker, labeled tubulin and yielded a diffuse broad band (Fig. 6B). These results suggested that probe 41 detected tubulin at a higher resolution than probe 40. The observed differences in resolution probably arose from the biotin–avidin detection system, which displayed better recognition between the avidin and biotin in biotin conjugates formed using a longer linker. The avidin-binding affinity (Kd value) of the biotinylated probe 41 was lower than that of the biotinylated probe 40. The identities of the two bands were investigated using immunoblot analysis with α- and β-specific antibodies. The upper and lower bands were found to correspond to the α- and β-tubulins, respectively (data not shown), suggesting that chemical probe 41 photolabeled both the α- and β-tubulin subunits.
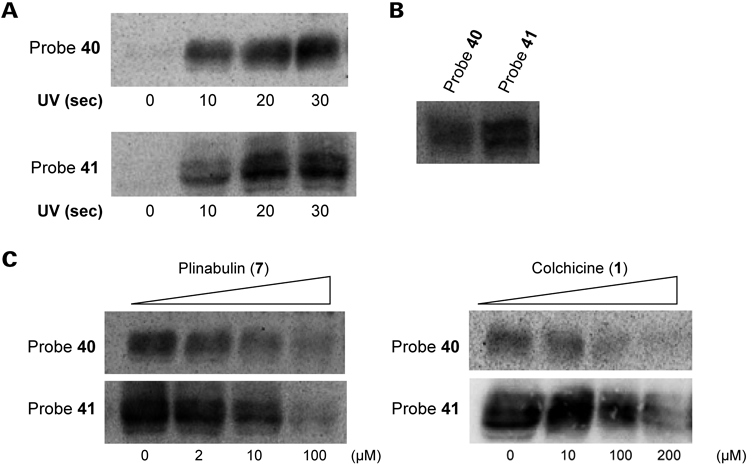

The photolabel specificity was examined using a competitive photoaffinity-labeling assay carried out in the presence or absence of plinabulin (7) and colchicine (1). Dose-dependent inhibition of the photoaffinity-labeling reaction was observed in the presence of compound 7 (Figs. 6C, 7B), indicating that probes 40–42 recognized the same binding site as compound 7. Photoaffinity labeling by probes 40–42 was also inhibited dose dependently (5–100 equiv.) in the presence of colchicine (Figs. 6C, 7B) but not in the presence of D-biotin as a negative control (data not shown). These results suggested that the synthetic probes 40–42 were effective as chemical probes. The recognition sites of the probes were proximally located around the intradimer region between the α- and β-tubulin subunits and were at or near the colchicine-binding site. The benzophenone unit of each chemical probe could photochemically modify either subunit.
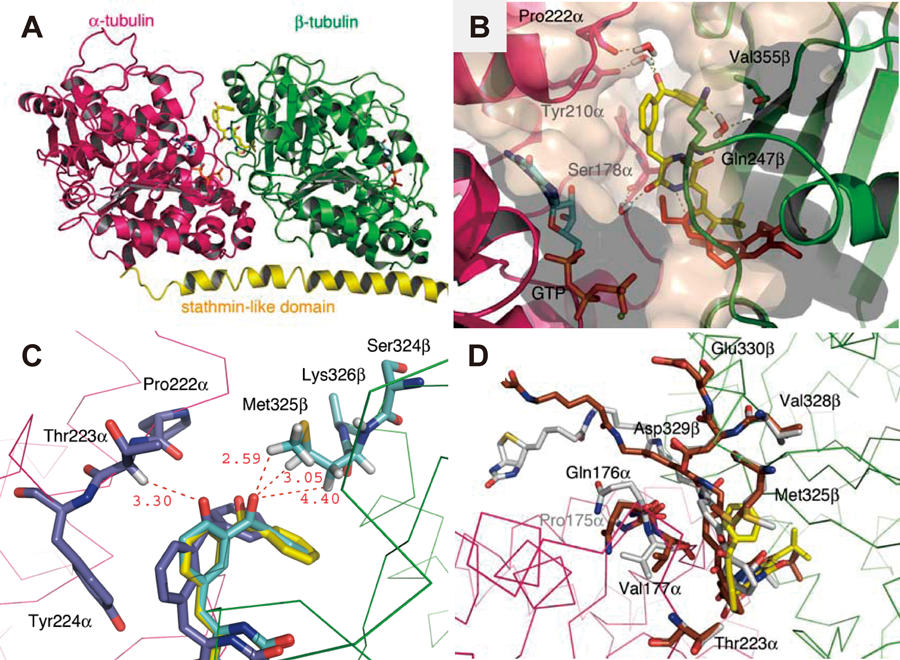
4.4. Molecular Modeling and Putative Binding MechanismThe binding modes of the plinabulin derivatives at the colchicine-binding site were further examined in view of the results of the tubulin photoaffinity-labeling study, as described in Section 4.3, and the results of a docking study conducted using the molecular modeling package MOE 2008.10 (Chemical Computing Group, Inc., Montreal, Canada). The calculations were simplified using the highly potent plinabulin derivative 39 in the docking study in place of the biotinylated probes. The tubulin proteins (chains α and β) were modeled based on the available crystallographic data for the complex with colchicine (PDB ID, 1SA0).49) Compound 39 was docked around the colchicine-binding site on the tubulin dimer, and the conformations with high docking scores were selected for examination. Energy minimization calculations and molecular dynamic (MD) simulations were then performed.
The docking of compound 39 into the colchicine-binding pocket on the β-tubulin subunit positioned the benzophenone oxygen of compound 39 far from the α-subunit, with a distance exceeding 7 Å (data not shown). In this case, it was difficult to photolabel both subunits. This result was not consistent with the results of the photoaffinity-labeling study, in which the chemical probe was observed to photolabel both the α- and β-tubulin subunits; however, docking of compound 39 to the outside of the colchicine-binding pocket, as shown in Fig. 8A, increased the likelihood of interactions with tubulin in a manner consistent with the experimental results. In this model, compound 39 was positioned at the interfacial region between the α- and β-subunits, and this region partially overlapped with the colchicine-binding pocket. Compound 39 interacted with Ser178α, Tyr210α, Pro222α, Val355β, and Gln247β on the α- and β-tubulins via four water molecules (Fig. 8B). A photoaffinity-labeling study using benzophenone revealed three accessible C–H bonds on the α-carbon of Thr223α and on the γ- and ε-carbons of Met325β. The distance between each carbonyl oxygen and hydrogen atom was less than 3.3 Å (Fig. 8C). The results from previous studies indicated that the reactive volume of the benzophenone moiety could be approximated as a sphere with a radius of 3.1 Å centered on the oxygen. Benzophenone selectively interacted with the Met residues with a photoaffinity-labeling radius of 6–7 Å.43) A modeling study involving probes 40 and 41, which included a long linker extending from the benzophenone moiety to the biotin label, suggested that the linker protruded from the interfacial region outside the molecule through a space formed between the α- and β-tubulin units. In this modeling study, the corresponding binding conformation of compound 39 on probe 41 was almost maintained (Fig. 8D). In addition, this modeling also suggested that the biotin linker on probe 42 protruded from the interfacial region away from the tubulin molecule through the interfacial space formed between the α- and β-subunits (data not shown). These results suggested the binding model shown in Fig. 8, offering a reasonable model for the photolabeling process involving both the α- and β-subunits. The plinabulin derivatives probably interact with the boundary region between the α- and β-tubulins around the colchicine-binding site and not inside the colchicine-binding site. The results of this docking study identified certain amino acid residues modified by the photoaffinity probe that would be of interest in the near future. Mass spectrometric analysis of the tubulin photoaffinity-labeling products is in progress.
5. Development of a Water-Soluble Prodrug of Plinabulin
Plinabulin (7) exhibited potent antitumor activity19) and an interesting binding mode36–38); however, its low water solubility (<0.1 µg/mL) needed to be improved to increase its pharmaceutical value. Compound 7 is currently injected in combination with a solubilizing agent that can induce hypersensitivity in the context of long-term administration. Therefore, we attempted to develop a water-soluble prodrug of 7 to avoid the safety or quality of life risks to patients.50,51)
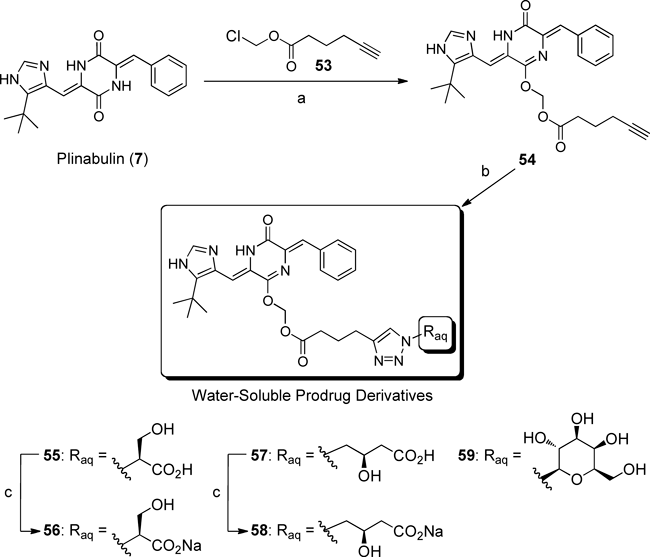
5.1. Design and Synthesis of a Water-Soluble Prodrug of PlinabulinThe development of an efficient synthetic approach to the water-soluble prodrug of 7 was complicated by the need to avoid transformations involving the mother skeleton. Therefore, the aqueous substituent, which contained unprotected polar functional groups, needed to be introduced directly. After repeated investigations, an effective strategy was designed involving an esterase hydrolysis via click chemistry.52) A copper-catalyzed alkyne azide coupling (CuAAC) reaction was suitable for achieving high chemoselectivity and a high yield under mild conditions using an unprotected water-solubilizing moiety.53)
Our strategy is illustrated in Chart 2. The C12 amide group of 7 was O-alkylated using the alkynoate 43, and then the alkyne part of 44 was reacted with the azide-type aqueous substituent 45 using CuAAC. The ester moiety of 46 was hydrolyzed by esterase to reproduce the parent compound 7. The resulting formaldehyde was expected to be converted promptly into carbon dioxide under the innate metabolic activity and exhaled, as in the pivoxil series.54,55)

Chart 2. Strategy for Preparing the Water-Soluble Prodrug of Plinabulin (7) Using Click Chemistry
Three types of polar group were examined as potential water-solubilizing moieties: 50–52. These groups were derived from the natural amino acids 47 or 48, or from the monosaccharide 49 (Fig. 9). The serine derivative 50 and the carnitine derivative 51 possessed β-hydroxycarboxylic acid moieties. These unprotected polar functional groups were similar; however, the structural differences that distinguished the β-amino acid from the γ-amino acid (the main difference involved the number of carbon atoms present) were expected to produce interesting effects on the water solubility and half-life properties of the prodrugs. 1-Azide-1-deoxy-β-D-galactopyranoside (52), which contained four hydroxyl groups, was predicted to be effective based on the high water solubility of glycosides in other natural products (Fig. 9).
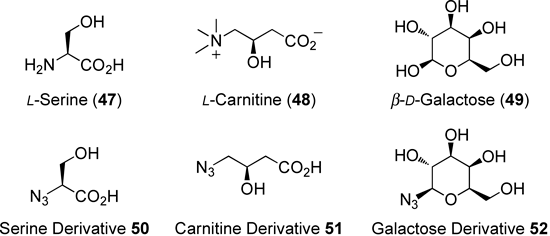
Fig. 9. Structures of the Natural Products 47–49 and the Azide Derivatives 50–52
Chart 3 shows that plinabulin (7) was successfully O-alkylated with the linker 53 using Cs2CO3, and the skeletally transformed compound 54 was obtained selectively. The structure of 54 was determined using X-ray crystallography.56) The monolactim 54 was subsequently connected to the azide-functionalized 50, 51, or 52 by CuAAC in the presence of CuSO4·5H2O and sodium ascorbate in H2O/tBuOH/DMF under microwave irradiation. These reactions proceeded without side reactions, and the triazole derivatives 55, 57, and 59 were obtained after HPLC purification. The carboxylic acid groups on the triazoles 55 and 57 were converted to the corresponding sodium salts 56 and 58 in the presence of a methacrylic acid ion-exchange resin, Diaion WK11 (Chart 3). Our original synthetic strategy for preparing water-soluble prodrug derivatives worked effectively and reproducibly, regardless of the identity of the azide substrate.
5.2. Evaluation of Water Solubility and in Vitro Hydrolysis by the EsteraseThe water solubility properties of 55–59 were examined. Compounds 55 and 57 dissolved slightly in water; however, the solubility could not be detected by HPLC. By contrast, the sodium salts of 56 and 58 were soluble in water, with solubility of 6.38 and 0.85 mg/mL, respectively. These values were, respectively, more than 64000-fold and 8500-fold greater than the solubility of plinabulin (7) (Table 7). These results indicated that the conversion to the sodium salt significantly enhanced water solubility. The moderate water solubility of 58 relative to 56 could be explained in terms of the difference in the relative hydrophobicities of the carnitine-type and serine-type water-solubilizing moieties.57) A slight increase in the hydrophobic properties, such as the number of carbons present, greatly influenced the water solubility of the prodrug. By contrast, the glycoside-type derivative 59 dissolved in water with a solubility of 0.0075 mg/mL. The water solubility of 59 was insignificant, and 59 was found to be unstable. These qualities of the glycoside-type derivative 59 resulted from the large number of hydroxyl groups, which modestly improved the water solubility, and the readily hydrolyzed ester moiety.
Table 7. Water Solubility and Half-lives of the Water-Soluble Prodrugs
56,
58, and
59| Compound | Water solubility (mg/mL) | Half-life (t1/2) (min) |
|---|
| 56 | 6.38 | 59.9 |
| 58 | 0.85 | 22.4 |
| 59 | 0.0075 | nta) |
| 7 [Plinabulin] | <0.1 µg/mL | nta) |
The utility of 56 and 58 as prodrugs was examined. Chart 4 shows that the synthesized 56 and 58 were dissolved in a PBS solution (pH 7.4) and hydrolyzed with esterase (porcine liver) at 37°C to generate the parent drug 7. The hydrolysates were analyzed using HPLC, which revealed that the hydrolysis reaction proceeded without the formation of any by-products, and only the parent drug 7 was produced. Esterase hydrolysis was confirmed to occur at the ester moiety because the absence of esterase in PBS gave only the starting materials. The half-life (t1/2) of 56 was 59.9 min, which is probably appropriate for water-soluble prodrug systems delivered by intravenous injection without a solubilizing agent. By contrast, the half-life (t1/2) of 58 was 22.4 min, only one-third the half-life of 56. This result indicated that the water-solubilizing moieties significantly influenced the esterase hydrolysis reaction. The bulkiness and stereochemistries of the auxiliaries, which were determined based on the natural amino acid groups, may have contributed to the hydrolysis rate.58–60) The influence of a simple change in the carbon chain length on the half-life may have been related to the hydrolysis mechanism of the esterase. Pig liver esterase, a serine hydrase, reacts with its substrates by forming an acyl-enzyme intermediate at the active site.61) Slight structural modifications in the carbon chain or stereochemistry at the water-solubilizing moiety could improve the recognition interaction with the parent drug.

The high water solubility and appropriate half-life of 56 indicated that the designed and synthesized candidate could potentially be useful as a water-soluble prodrug of plinabulin (7). Furthermore, the sodium salt 56 was stable over 1 month at 4°C without decomposition.
5.3. Evaluation of CytotoxicityThe cytotoxic activities of the water-soluble prodrug 56 and the water-soluble auxiliary 60 were verified in vitro using the human colon HT-29 cell line (Table 8). 56 exhibited an IC50 value that was one-tenth the value of 7 under normal conditions.62,63) The reduced IC50 value was attributed to the direct reaction of the monolactim prodrug 56 with small amounts of esterases produced by the cells or with the inner components of the cells. On the other hand, the water-soluble auxiliary 60 proved to be nontoxic, even at high concentrations. These results supported the likelihood that the water-soluble prodrug system should be safe and effective in humans.
Table 8. Cytotoxic Activity toward Human Colon HT-29 Cells
| Compound | IC50 (nM)/HT-29a) |
|---|
| 56 | 101.4±5.7 |
| 60 | >2 mM |
| 7 [Plinabulin] | 13.5±2.2 |
| Colchicine | 16.6±0.9 |
a) Values indicate the mean or mean±S.E.M. (for potent compounds) from at least three independent dose–response curves.
6. Conclusion
This review discusses the development process by which novel antimicrotubule agents with potent cytotoxicity and vascular-disrupting activity were identified based on the chemical structure of the natural DKP phenylahistin, which displayed colchicine-like tubulin-depolymerizing activity. Among the synthesized didehydroDKP derivatives, plinabulin (7) was selected as a clinical candidate for anticancer therapy. This molecule has been in phase II clinical trials as a VDA. Further SAR studies based on 7 identified more potent didehydroDKP derivatives having a substitution on the phenyl ring of compound 7. One such derivative, compound 38m, which features a benzophenone structure, exhibited highly potent cytotoxicity, with an IC50 value of 0.5 nM against HT-29 cells. The precise tubulin-binding mode of compound 7 was examined by developing biotin-tagged chemical probes possessing a photoreactive benzophenone structure in the pharmacophore, and a photoaffinity-labeling study was performed in the presence of tubulin. Although the binding site has not yet been elucidated in detail, compound 7 was found to recognize both the α- and β-tubulin subunits at the interfacial region located near or at the colchicine-binding site. The very low water solubility of compound 7 (<0.1 µg/mL, H2O) was improved by developing a prodrug strategy based on click chemistry, and a highly water-soluble prodrug 56 (6.38 mg/mL, H2O) with a serine-like water-solubilizing moiety was obtained. The prodrug 56 was converted to the parent 7 in vitro with a half-life of 59.9 min in the presence of porcine liver esterase under physiologic conditions. The studies conducted by our group have contributed significantly to the practical development of cancer therapeutics. Further studies for the development of more potent, effective anticancer DKP derivatives with novel chemical scaffolds and unique prodrug structures are underway, with the focus on developing a cancer-selective antibody. Our aim is to develop an understanding of the molecular mechanisms underlying plinabulin action. These studies will be reported elsewhere in the near future.
Acknowledgment
This research was partially carried out at Kyoto Pharmaceutical University. The authors are grateful to Prof. Yoshiaki Kiso, who provided the environment and opportunities for this research. The authors thank Prof. Hiroyuki Yasui, Prof. Miki Akamatsu, Dr. Takeo Usui, Dr. Kyoko Kohno, and Dr. Koushi Hidaka for their kind help and useful suggestions. This research was supported by Grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, including Grants-in-Aid for Scientific Research (B) 20390036 and 23390029, a Grant-in-Aid for Young Scientists (B) 23790143, and a Grant from the Frontier Research Program of MEXT. The drug development of plinabulin including preclinical and clinical studies was performed solely by Nereus Pharmaceuticals, Inc. (San Diego, CA, U.S.A.). We appreciate their efforts.
References
- 1) Perez E. A., Mol. Cancer Ther., 8, 2086–2095 (2009).
- 2) Dumontet C., Jordan M. A., Nat. Rev. Drug Discov., 9, 790–803 (2010).
- 3) Zhou J., Giannakakou P., Curr. Med. Chem. Anticancer Agents, 5, 65–71 (2005).
- 4) Pilat M. J., LoRusso M. J., J. Cell. Biochem., 99, 1021–1039 (2006).
- 5) Lin C. M., Ho H. H., Pettit G. R., Hamel E., Biochemistry, 28, 6984–6991 (1989).
- 6) Pettit G. R., Temple C. Jr., Narayanan V. L., Varma R., Simpson M. J., Boyd M. R., Rener G. A., Bansal N., Anticancer Drug Des., 10, 299–309 (1995).
- 7) Ohsumi K., Hatanaka T., Nakagawa R., Fukuda Y., Morinaga Y., Suga Y., Nihei Y., Ohishi K., Akiyama Y., Tsuji T., Anticancer Drug Des., 14, 539–548 (1999).
- 8) Koyanagi N., Nagasu T., Fujita F., Watanabe T., Tsukahara K., Funahashi Y., Fujita M., Taguchi T., Yoshino H., Kitoh K., Cancer Res., 54, 1702–1706 (1994).
- 9) Daenen L. G. M., Roodhart J. M. L., Shaked Y., Voest E. E., Curr. Clin. Pharmacol., 5, 178–185 (2010).
- 10) Kanoh K., Kohno S., Asari T., Harada T., Katada J., Muramatsu M., Kawashima H., Sekiya H., Uno I., Bioorg. Med. Chem. Lett., 7, 2847–2852 (1997).
- 11) Kanoh K., Kohno S., Katada J., Hayashi Y., Muramatsu M., Uno I., Biosci. Biotechnol. Biochem., 63, 1130–1133 (1999).
- 12) Kanoh K., Kohno S., Katada J., Takahashi J., Uno I., J. Antibiotics (Tokyo), 52, 134–141 (1999).
- 13) Nicholson B., Lloyd G. K., Miller B. R., Palladino M. A., Kiso Y., Hayashi Y., Neulteboom S. T. C., Anticancer Drugs, 17, 25–31 (2006).
- 14) Mita M. M., Spear M. A., Yee L. K., Mita A. C., Heath E. I., Papadopoulos K. P., Federico K. C., Reich S. D., Romero O., Malburg L., Pilat M., Lloyd G. K., Neuteboom S. T. C., Cropp G., Ashton E., LoRusso P. M., Clin. Cancer Res., 16, 5892–5899 (2010).
- 15) Ferrer E., Bolós J., Castañer R., Drugs Future, 35, 11–15 (2010).
- 16) Hayashi Y., Orikasa S., Tanaka K., Kanoh K., Kiso Y., J. Org. Chem., 65, 8402–8405 (2000).
- 17) Kanoh K., Kohno S., Katada J., Takahashi J., Uno I., Hayashi Y., Bioorg. Med. Chem., 7, 1451–1457 (1999).
- 18) Kanzaki H., Yanagisawa S., Kanoh K., Nitoda T., J. Antibiotics (Tokyo), 55, 1042–1047 (2002).
- 19) Yamazaki Y., Tanaka K., Nicholson B., Deyanat-Yazdi G., Potts B., Yoshida T., Oda A., Kitagawa T., Orikasa S., Kiso Y., Yasui H., Akamatsu M., Chinen T., Usui T., Shinozaki Y., Yakushiji F., Miller B. R., Neuteboom S., Palladino M., Kanoh K., Lloyd G. K., Hayashi Y., J. Med. Chem., 55, 1056–1071 (2012).
- 20) Yamazaki Y., Sumikura M., Masuda Y., Hayashi Y., Yasui H., Kiso Y., Chinen T., Usui T., Yakushiji F., Potts B., Neuteboom S., Palladino M., Lloyd G. K., Hayashi Y., Bioorg. Med. Chem., 20, 4279–4289 (2012).
- 21) Suzuki M., Iwasaki T., Miyoshi M., Okumura K., Matsumoto K., J. Org. Chem., 38, 3571–3575 (1973).
- 22) Weitman M., Lerman L., Cohen S., Nudelman A., Major D. T., Gottlieb H. E., Tetrahedron, 66, 1465–1471 (2010).
- 23) Bredereck H., Theilig G., Chem. Ber., 86, 88–96 (1952).
- 24) Folkes A., Roe M. B., Sohal S., Golec J., Faint R., Brooks T., Charlton P., Bioorg. Med. Chem. Lett., 11, 2589–2592 (2001).
- 25) Gallina C., Liberatori A., Tetrahedron, 30, 667–673 (1974).
- 26) Böhm H.-J., Banner D., Bendels S., Kansy M., Kuhn B., Müller K., Obst-Sander U., Stahl M., ChemBioChem, 5, 637–643 (2004).
- 27) Pettit G. R., Toki B., Herald D. L., Verdier-Pinard P., Boyd M. R., Hamel E., Pettit R. K., J. Med. Chem., 41, 1688–1695 (1998).
- 28) Ghinet A., Rigo B., Hénichart J.-P., Broc-Ryckewaert D. L., Pommery J., Pommery N., Thuru X., Quesnel B., Gautret P., Bioorg. Med. Chem., 19, 6042–6054 (2011).
- 29) Liou J.-P., Chang J.-Y., Chang C.-W., Chang C.-Y., Mahindroo N., Kuo F.-M., Hsieh H.-P., J. Med. Chem., 47, 2897–2905 (2004).
- 30) Hsieh H.-P., Liou J.-P., Lin Y.-T., Mahindroo N., Chang J.-Y., Yang Y.-N., Chern S.-S., Tan U.-K., Chang C.-W., Chen T.-W., Lin C.-H., Chang Y.-Y., Wang C.-C., Bioorg. Med. Chem. Lett., 13, 101–105 (2003).
- 31) Andreu J. M., Perez-Ramirez B., Gorbunoff M. J., Ayala D., Timasheff S. N., Biochemistry, 37, 8356–8368 (1998).
- 32) Nam N.-H., Kim Y., You Y.-J., Hong D.-H., Kim H.-M., Ahn B.-Z., Bioorg. Med. Chem. Lett., 12, 1955–1958 (2002).
- 33) Zuse A., Schmidt P., Baasner S., Böhm K. J., Müller K., Gerlach M., Günther E. G., Unger E., Prinz H., J. Med. Chem., 50, 6059–6066 (2007).
- 34) Álvarez C., Álvarez R., Corchete P., Pérez-Melero C., Peláez R., Medarde M., Bioorg. Med. Chem., 16, 8999–9008 (2008).
- 35) Andreu J. M., Timasheff S. N., Biochemistry, 21, 6465–6476 (1982).
- 36) Yamazaki Y., Kohno K., Yasui H., Kiso Y., Akamatsu M., Nicholson B., Deyanat-Yazdi G., Neuteboom S., Potts B., Lloyd G. K., Hayashi Y., ChemBioChem, 9, 3074–3081 (2008).
- 37) Yamazaki Y., Sumikura M., Hidaka K., Yasui H., Kiso Y., Yakushiji F., Hayashi Y., Bioorg. Med. Chem., 18, 3169–3174 (2010).
- 38) Yamazaki Y., Kido Y., Hidaka K., Yasui H., Kiso Y., Yakushiji F., Hayashi Y., Bioorg. Med. Chem., 19, 595–602 (2011).
- 39) Hashimoto M., Hatanaka Y., Eur. J. Org. Chem., 2008, 2513–2523 (2008).
- 40) Kotzyba-Hibert F., Kapfer I., Goeldner M., Angew. Chem. Int. Ed. Engl., 34, 1296–1312 (1995).
- 41) Dormán G., Prestwich G. D., Trends Biotechnol., 18, 64–77 (2000).
- 42) Dormán G., Prestwich G. D., Biochemistry, 33, 5661–5673 (1994).
- 43) Vodovozova E. L., Biochemistry (Moscow), 72, 1–20 (2007).
- 44) Hofmann K., Finn F. M., Kiso Y., J. Am. Chem. Soc., 100, 3585–3590 (1978).
- 45) Hofmann K., Kiso Y., Proc. Natl. Acad. Sci. U.S.A., 73, 3516–3518 (1976).
- 46) Sato S., Kwon Y., Kamisuki S., Srivastava N., Mao Q., Kawazoe Y., Uesugi M., J. Am. Chem. Soc., 129, 873–880 (2007).
- 47) Saito S., Prog. Mol. Subcell. Biol., 46, 187–219 (2009).
- 48) Nunes M., Kaplan J., Wooters J., Hari M., Minnick A. A. Jr., May M. K., Shi C., Musto S., Beyer C., Krishnamurthy G., Qiu Y., Loganzo F., Ayral–Kaloustian S., Zask A., Greenberger L. M., Biochemistry, 44, 6844–6857 (2005).
- 49) Ravelli R. B., Gigant B., Curmi P. A., Jourdain I., Lachkar S., Sobel A., Knossow M., Nature (London), 428, 198–202 (2004).
- 50) Yakushiji F., Tanaka H., Muguruma K., Iwahashi T., Yamazaki Y., Hayashi Y., Chemistry, 17, 12587–12590 (2011).
- 51) Yakushiji F., Tanaka H., Muguruma K., Iwahashi T., Yamazaki Y., Hayashi Y., Chem. Pharm. Bull., 60, 877–881 (2012).
- 52) Kolb H. C., Finn M. G., Sharpless K. B., Angew. Chem. Int. Ed., 40, 2004–2021 (2001).
- 53) Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. Int. Ed., 41, 2596–2599 (2002).
- 54) Totsuka K., Shimizu K., Konishi M., Yamamoto S., Antimicrob. Agents Chemother., 36, 757–761 (1992).
- 55) Nakashima M., Uematsu T., Oguma T., Yoshida T., Mizojiri K., Matsuno S., Yamamoto S., Antimicrob. Agents Chemother., 36, 762–768 (1992).
- 56) CCDC, 829268 contains the supplementary crystallographic data for 54. These data may be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 57) Chalikian T. V., J. Phys. Chem. B, 105, 12566–12578 (2001).
- 58) Jiang Y., Hu L., Bioorg. Med. Chem. Lett., 17, 517–521 (2007).
- 59) Foulds G., Knirsch A. K., Lazar J. D., Tensfelt T. G., Gerber N., Antimicrob. Agents Chemother., 35, 665–671 (1991).
- 60) Ahmed S., Imai T., Otagiri M., Pharm. Res., 13, 1524–1529 (1996).
- 61) Tamm C., Pure Appl. Chem., 64, 1187–1191 (1992).
- 62) Nam N.-H., Kim Y., You Y.-J., Hong D.-H., Kim H.-M., Ahn B.-Z., Bioorg. Med. Chem., 11, 1021–1029 (2003).
- 63) Fürst R., Zupkó I., Berényi A., Ecker G. F., Rinner U., Bioorg. Med. Chem. Lett., 19, 6948–6951 (2009).
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/FT1.png%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/FT2.png%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)
;%0A%09%09%09newWindow.document.open();%0A%09%09%09newWindow.document.write('<img src=%22./Graphics/FT3.png%22>');%0A%09%09%09newWindow.document.close();%0A%09%09)