Review
In Situ Protection Methodology in Carbonyl Chemistry
2014 年 62 巻 1 号 p. 1-11
詳細
2014 年 62 巻 1 号 p. 1-11
Recent progress in selective transformation of carbonyl groups using in situ protection methodology is described. These techniques which enable reversing reactivity of functional groups have potential usefulness because they can remove complicated protection–deprotection sequences. In this review, we discuss various in situ protection strategies and their synthetic applications.
Carbonyl groups, and especially their characteristic C–C and C–H bond forming reactions, play key roles in organic chemistry. It is well known that the order of reactivity of carbonyl groups toward nucleophiles is aldehydes>ketones> esters (Chart 1). This reactivity pattern has been used advantageously to develop a variety of processes for chemoselective reactions of the most reactive carbonyl groups in substrates which contain multiple C=O moieties.1) In contrast, the design of reactions that target less reactive carbonyl groups in substances of this type is a much more difficult task. For this purpose, protective group strategies are needed, as is exemplified by the general three-step, protection/reaction/deprotection sequence required to promote ketone selective reactions of keto-aldehydes (Chart 2).


In 1979, Luche and Gemal described an in situ protection methodology,2,3) in which a one-pot operation is used to install a protective group that is then removed following reaction at another center (Chart 3). This new technology, enabling the design of selective, one-pot transformations of less reactive carbonyl groups in the presence of more reactive counterparts, is the topic of the review given below.

The first in situ protection method described by Luche and Gemal is the selective reduction of ketones in the presence of aldehydes. Thus, the addition of 1 molar eq of CeCl3 hexahydrate to the equimolar mixture of aldehyde and ketone or substrates that contain aldehyde and ketone groups can yield hydrate of aldehydes selectively. The addition of NaBH4 reduces the remaining ketones selectively, and usual work-up affords the hydroxy aldehyde products2,3) (Chart 4, Table 1). Although simple to perform and selective in generating reduced ketone products in high yields (Runs 1–3, 6, 7), this procedure is not applicable to selective reactions of ketone substrates in the presence of conjugated and aromatic aldehydes, which do not readily form hydrates (Runs 4, 5).

| Run | RCHO/R′COR″ | Yield of reductants (%) |
|---|---|---|
| 1 | Hexanal | 2 |
| Cyclohexanone | 100 | |
| 2 | Hexanal | 13 |
| 2-Octanone | 96 | |
| 3 | Cyclohexanecarboxaldehyde | 22 |
| Phenylacetone | 93 | |
| 4 | Benzaldehyde | 60 |
| Acetophenone | 100 | |
| 5 | Citral | 70 |
| 2-Cyclohexenone | 100 | |
| 6 |  | 95(Hydroxy aldehyde) |
| 7 |  | 85(Hydroxy aldehyde) |
After the report by Luche, a large number of investigations uncovered various other types of in situ protection methodologies, which enable chemoselective reduction, alkylation, and even Wittig reactions of carbonyl substrates.
For example, in 1980 Paradisi et al. devised an aldimine protection protocol, which had been employed previously in steroidal dicarbonyl compounds,4,5) to carry out one-pot reductions of ketones in the presence of aldehydes and in substrates that also contain aldehyde groups6,7) (Chart 5, Table 2). In this approach, the aldehyde is selectively protected by reaction with tert-butylamine that produces an aldimine in situ. Reduction of the remaining ketone moiety with LiAlH(Ot-Bu)3 is then followed by aqueous acid work-up which transforms the aldimine to the original aldehyde group (Table 2). Although this procedure is applicable to the protection of conjugated and aromatic aldehydes (Runs 3, 4), it cannot be used to conduct selective reductions of conjugated ketones owing to the occurrence of competitive conjugate reduction. In addition, Paradisi showed that this method can be utilized to perform ketone selective Wittig methylenation reactions (Runs 6, 8, 9).8)

| Run | Initial mixture | Reagent | Recovered starting material (%) | Product (%) |
|---|---|---|---|---|
| 1 | Octanal | LiAlH(Ot-Bu)3 | 98 | 1 |
| 2-Heptanone | 0 | 100 | ||
| 2 | Octanal | LiAlH(Ot-Bu)3 | 97 | 2 |
| Cyclohexanone | 0 | 100 | ||
| 3 | Benzaldehyde | LiAlH(Ot-Bu)3 | 97 | <1 |
| 2-Heptanone | 0 | 100 | ||
| 4 | Geranial | LiAlH(Ot-Bu)3 | 83 | <1 |
| Acetophenone | 0 | 99 | ||
| 5 |  | LiAlH(Ot-Bu)3 | 89(β-OH : α-OH=83/6) | |
| 6 | H2C=PPh3 | 85 | ||
| 7 |  | LiAlH(Ot-Bu)3 | 2 | 89(β-OH : α-OH=84/5) |
| 8 | H2C=PPh3 | 66 | ||
| 9 |  | H2C=PPh3 | 84 |
A method for chemoselective transformations of substances containing aldehyde groups and other reactive centers that relies on in situ formation of N,O-acetal intermediates, which are prepared by the reaction of lithium amides and aldehydes, and stable to further reaction of organometallic reagents, has been devised. In 1981, Comins et al. designed a strategy that utilizes lithium morpholide and lithium 2-(N-methyl-N-(2-pyridyl))amide (LMPA) as aldehyde protecting reagents9) (Table 3). The scope of the use of lithium morpholide is limited to non-enolizable substrates such as aromatic ones due to its strong basic properties. For example, it reacts with esters to form enolates. On the other hand, LMPA can be utilized to protect aldehydes selectively even in the presence of enolizable ester moieties. In the in situ protection strategy, selective addition of LMPA to the aldehyde moiety takes place to form adduct 1 (Chart 6) that is stable to Grignard reagents at below 0°C. Although adduct 1 is susceptible to organolithium reagents and LiAlH4, the trimethylsilyl (TMS) protected LMPA adduct 2 is stable even to such reagents. Following reaction at the other reactive center, the protective groups exemplified by 1 and 2 are quantitatively removed by hydrolysis using dilute HCl (Chart 6). Comins demonstrated the utility of this protection method by applying it to a variety of chemical reactions of aldehyde containing multi-functional substrates (Table 3), including aryl bromide halogen-metal exchange (Runs 1, 4), ester alkylation and reduction (Runs 2, 3), and nitrile α-benzylation (Run 5). The LMPA methodology is only applicable to protection of aldehydes and not ketones because the latter (e.g. acetophenone, benzylacetone, and 4-tert-butylcyclohexanone) are converted to corresponding enolate anions via reaction with this basic reagent. The use of other lithium amides for in situ protection via the formation of N,O-acetal intermediates has been previously reviewed10) and will not be discussed in detail in this review.
| Run | Reactant(s) | Reaction conditions | Product(s) | Yield (%) |
|---|---|---|---|---|
| 1 | 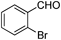 | 1. Lithium morpholide, −78°C |  | 85 |
| 2. nBuLi, −78°C | ||||
| 3. MeI | ||||
| 2 |  | 1. LMPA, −23°C |  | 87/90 |
| 2. MeMgCl | ||||
| 3 |  | 1. LMPA, −23°C |  | 89 |
| 2. TMSCl | ||||
| 3. LiAlH4 | ||||
| 4 |  | 1. LMPA, −78°C |  | 86 |
| 2. TMSCl, THF | ||||
| 3. nBuLi | ||||
| 4. MeI | ||||
| 5 |  | 1. LMPA, −78°C |  | 80/71 |
| 2. TMSCl | ||||
| 3. LDA | ||||
| 4. BnCl |

Lambert and colleagues applied the Comins’ in situ protection method in a sequence for the synthetic study of a taxol-like oxetan, and a ketone selectively alkylated aldehyde was obtained from keto aldehyde in moderate yield11) (Chart 7).

Roschangar et al. also developed an aldehyde protection strategy that utilizes lithium N,O-dimethylhydroxylamide (LiN(OMe)Me) as an alternative in situ protecting reagent with low ortho-directing property.12) An example of the use of this methodology is found in the route shown in Chart 8, in which in situ aryl aldehyde protection proceeds formation of a key boronate that is employed in a direct Suzuki–Miyaura coupling reaction to afford the drug candidate (Chart 8).

As metal amides, in 1969 Lappert and colleagues described the reaction of cinnamaldehyde with Ti(NMe2)4 that produces the corresponding gem-diamine (aminal) via 1,2-addition.13) In 1982, Reetz and Wenderoth observed that allyl titanium ate complexes bearing amine ligands, which were prepared by the reaction of allyllithium and titanium tetraamide, can be employed to accomplish ketone selective allylation reactions in the presence of aldehydes. The type of ligand is very important. Titanium ate complexes with amino ligands showed ketone selective allylation whereas the one with alkoxy ligand showed complete reversal chemoselectivity14) (Chart 9). Following the initial report, they discovered that the use of 1 eq of titanium tetrakis(diethylamide) serves as an excellent reagent for in situ protection of aldehydes to give N,O-acetals, which readily revert to aldehydes upon aqueous work-up, in the presence of ketones15) (Chart 10) and can be used to carry out selective alkylation reactions of ketones in the presence of aldehydes with organolithium reagents, including the lithium enolate derived from ethyl acetate (Table 4). For ketone–ketone discrimination, titanium tetrakis(diethylamide) does not work well because the reagent needs higher reaction temperatures (ca. −30°C) to form the adducts and at which temperature they begin to fragment. In such cases, titanium reagents containing N,N-dimethylamine ligands are able to discriminate between the ketone carbonyl groups based on steric factors, although the reagent is more reactive and reacts with both aldehydes and ketones (Chart 11). A limitation of this methodology comes from the fact that only very reactive carbon nucleophiles can be used in reactions that follow the in situ protection steps because reactions need to be conducted at temperatures below −30°C in order to maintain the stability of the protected form.


 | ||
|---|---|---|
| Run | Reagent | Ratio of yield (A : B) (%) |
| 1 | MeLi | <1 : >99 |
| 2 | nBuLi | <1 : >99 |
| 3 |  | <1 : >99 |

Metal amide chemistry was extended to other systems by Hosomi et al. Thus, titanium amides such as Ti(NEt2)4 readily undergo Michael additions to α,β-unsaturated esters and ketones, which give intermediate tris(amino)titanium enolates that can be intercepted with aldehydes. The overall process corresponds to a tandem conjugate addition-aldol reaction sequence.16)
4.3. Aluminum amideIn 1988, Yamamoto and colleagues described amphiphilic methodology that uses aluminum reagents and organolithium reagents17) (Chart 12, Eq. 1). They first found that aldehyde carbonyls are electrophilically activated by methyl aluminum bis(2,6-di-tert-butyl-4-methylphenoxide) (MAD) (structure is shown in Chart 20) to afford the alkylation products of aldehydes exclusively in the presence of ketones. They hypothesized that an organoaluminum reagent of type R2AlX (X: hetero atom) reacts with aldehydes faster than ketones to form acetal type intermediates which are inert to nucleophiles. An investigation of various aluminum reagents, including aluminum phenoxide, thiolates, and amides, demonstrated that the aluminum complex bearing two methyl and one N-methylanilide group, Me2AlNMePh, is ideal for carrying out ketone selective alkylation reactions in mixtures containing aldehydes (Chart 12, Eq. 2) (Table 5).
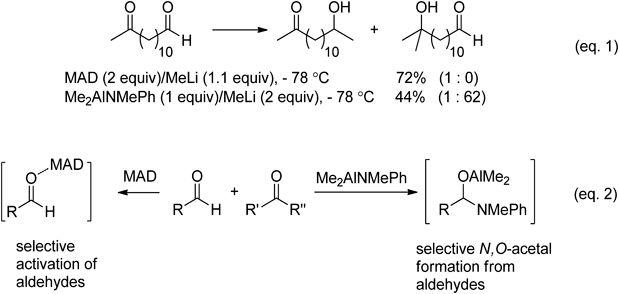
 | ||||
|---|---|---|---|---|
| Run | Substrates | R′Li | Products | Yield (A/B) (%) |
| 1 |  | MeLi |  | 13/92 |
| 2 |  | MeLi |  | 9/83 |
| 3 | PhLi | 7/100 | ||
| 4 |  | MeLi |  | 12/70 |
| 5 | PhLi | 7/97 | ||
Colby and colleagues recently reported that the aluminum complex prepared from diisobutylaluminum hydride (DIBAL-H) and N,O-dimethylhydroxylamine (Weinreb amine) serves as a versatile reagent to mask more reactive carbonyl groups as N,O-acetals, which are inert to organolithiums, Grignard reagents, and borohydrides, in the presence of lesser reactive counterparts. Equation 1 of Chart 13 shows their strategy, and as optimization of conditions addition of i-PrMgCl before adding nucleophile gave the best result18,19) (Chart 13, Eq. 2). They demonstrated that selective alkylation, reduction, and Wittig methylenation reactions of ketones and esters can be carried out in the presence of aldehydes and alkylations and reductions of esters in the presence of ketones can be performed by using this in situ protection methodology (Table 6). It is noteworthy that the method was applied to lactone selective alkylation reaction of α-santonin, which contains a highly reactive dienone moiety (Run 8). Because the aluminum amide reagent reacts with both aldehyde and ketone groups, albeit at different rates, its use in discriminating between these carbonyl moieties requires strict control of the stoichiometry of the reagent.

| Run | Substrate | Reagent (equiv) | Product | Yield (%) |
|---|---|---|---|---|
| 1 |  | MeLi (2) |  | 63 |
| 2 | LiBEt3H (1) |  | 86 | |
| 3 |  |  | 70 | |
| 4 | H2C=PPh3 (2.5) |  | 80 | |
| 5 |  | MeLi (2) |  | 65 |
| 6 |  | MeMgBr (6.8) |  | 70 |
| 7 | LiBEt3H (5) |  | 78 | |
| 8 | 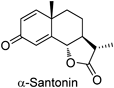 | MeLi (3.6) |  | 69 |
Recently, Marko and colleagues showed that 1.1 eq of diethylaluminum benzenethiolate reacts with aldehydes selectively to form O,S-acetals in the presence of ketones at −78°C (Chart 14), processes that in situ protect against DIBAL-H reduction.20) This reaction was applied to the selective reduction of ketones and esters in the presence of aldehydes (Table 7). Although the aluminum thiolate reagent partially reacts with ketone to form corresponding ketal intermediates, by using careful control of reaction conditions it can be employed for the selective protection of aldehydes.

 | ||||
|---|---|---|---|---|
| Run | Substrate | Product | Selectivity (%) | Yield (%) |
| 1 |  |  | 98 | 89 |
| 2 |  |  | 91 | 82 |
| 3 |  |  | >99 | 91 |
| 4 |  | 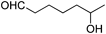 | 92 | 89 |
| 5 |  | 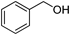 | >99 | 97 |
| 6 |  |  | >99 | 93 |
Although several methods are available for selective formation of acetals from aldehydes in the presence of ketones, selective ketal formations from ketones in the presence of aldehydes are difficult to execute.21) In addition, only one method exists for selective deprotection of acetals in the presence of ketals.22,23) In 1992, Kim et al. described a ketone selective dioxolanation reaction that takes place in the presence of an aldehyde.24) A combination of dimethyl sulfide and trimethylsilyl trifluoromethanesulfonate (TMSOTf) (Chart 15) was used to selectively protect aldehydes by formation of the corresponding O,S-acetal type sulfonium salts in the presence of ketones, and after carrying out selective ketalization of the remaining ketones using Noyori’s conditions, rebirth of aldehydes with alkaline work-up gives ketal aldehydes25) (Table 8). However, O,S-acetal type sulfonium salts react with nucleophiles. Thus, it is possible to selectively promote allylation reactions of aldehydes in the presence of ketones by first forming O,S-acetal type intermediates.26) In addition, reactions of α,β-unsaturated ketones with Me2S-TMSOTf produces Michael adducts that react with various nucleophiles.27)

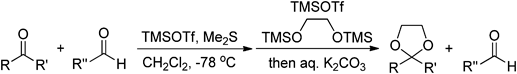 | |||||||
|---|---|---|---|---|---|---|---|
| Run | Substrates | Yield (%) | Run | Substrate | Product | Yield (%) | |
| Ketal | Aldehyde | ||||||
| 1 |  | 99 | 96 | 4 |  |  | 80 |
| 2 |  | 99 | 99 | 5 |  |  | 65 |
| 3 |  | 96 | 96 | 6 |  |  | 66 |
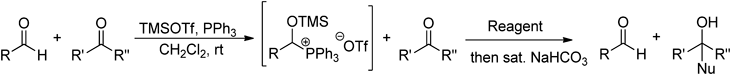
Recently, we found that the reactivity of the phosphonium salts is strongly governed by the kind of phosphine.28) Thus, in the Grignard reaction of the phosphonium salts obtained by the reaction of acetals and phosphines, phosphonium salts from Tris-o-tolyl phosphine (P(o-Tol)3) are active whereas the phosphonium salts from PPh3 are somewhat less reactive (Chart 16). We then applied this phenomena to the substrates having two different carbonyl groups and developed a technique for selective transformations of ketones and esters in the presence of aldehydes that utilizes pretreatment with a combination of PPh3 and TMSOTf29) (Table 9). The in situ protection method, which proceeds via O,P-acetal type phosphonium salt intermediates,30) is applicable to performing alkylation reactions with Grignard reagents and the reduction reactions with borane or DIBAL-H. The combination of PPh3 and TMSOTf converts only aldehydes and not ketones nor esters to corresponding O,P-acetal type phosphonium salts even when excess amounts of the reagents are used (Chart 17). On the other hand, by changing the phosphine from PPh3 to PEt3, O,P-ketal type phosphonium salt formation takes place with ketones (Chart 18). As a result, selective transformations of esters in the presence of ketones can be accomplished (Table 10). It is noteworthy that tert-butyldimethylsilyl (TBS) and methoxymethyl (MOM) group in substrates survive the in situ protection/deprotection processes (Runs 5, 6). We have utilized this protection strategy for ketone selective Corey–Bakshi–Shibata (CBS) reduction in a highly efficient 5-step asymmetric total synthesis of (+)-centrolobine (Chart 19). To our knowledge, this is the first and only observation of asymmetric transformation of a ketone in the presence of an aldehyde.

 | ||||
|---|---|---|---|---|
| Run | Substrate | Reagent | Product | Yield (%) |
| 1 |  | BH3·THF |  | 96 |
| 2 | PhMgBr | 93 | ||
| 3 | EtMgCl | 87 | ||
| 4 |  | BH3·THF |  | 87 |
| 5 |  | BH3·THF | 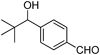 | 74 |
| 6 |  | BH3·THF |  | 87 |
| 7 |  | DIBAL-H |  | 80 |
| 8 | EtMgCl | 76 | ||

 | ||||
|---|---|---|---|---|
| Run | Substrate | Reagent | Product | Yield (%) |
| 1 |  | DIBAL-H |  | 82 |
| 2 |  | DIBAL-H |  | 76 |
| 3 |  | DIBAL-H |  | 93 |
| 4 | MeMgBr | 83 | ||
| 5 |  | EtMgCl |  | 80 |
| 6 | 74 | |||

A combination of PPh3 and silyltriflate was also reported to participate in conjugate addition reactions of α,β-unsaturated ketones to give other phosphonium salts, which serve as useful intermediates for the construction of β-substituted enones using Wittig olefination reactions.31)
In 1988, Yamamoto and colleagues described the use of the highly bulky aluminum complex, methyl aluminum bis(2,6-di-tert-butyl-4-methylphenoxide (MAD, Chart 20, upside left), that displays steric and electronic based discriminant complex formation with ketones. As a result, this Lewis acid can be used to carry out selective reduction reactions of sterically different ketones32) (Table 11). In each of these reactions, the bulky aluminum reagent forms a complex with a less hindered or more electrically labile carbonyl group, which effectively blocks reaction with the nucleophilic reducing agent (Chart 20, Eq. 1). It is interesting that this Lewis acid operates as an inactivator and not as an activator of carbonyl groups. In reactions of some aromatic ketones, the use of Br2AlH, prepared from LiAlH4 and 3 equiv of AlBr3, rather than DIBAL-H was found to be superior (Run 2). Although this method effectively discriminates ketones, it is not applicable for carrying out selective reactions of ketones in the presence of aldehydes owing to the high reactivity of an aldehyde-MAD complex. In 1993, however, Yamamoto and colleagues observed that methylaluminum bis-(2,6-diphenylphenoxide) (MAPH) (Chart 20, upside right) is a useful agent for promoting selective addition of organolithium reagents to more hindered aldehydes and ketones in the presence of less hindered aldehydes33) (Table 12).

| Run | Ketones | MAD (equiv) | Hydride (equiv) | Combined yield of reduced alcohols (%) | Ratio |
|---|---|---|---|---|---|
| 1 |  | 1 | DIBAL-H (1) | 79 | 1/2.6 |
| 2 | 2 | Br2AIH (2) | 68 | 1/6 | |
| 3 |  | 1 | DIBAL-H (1) | 66 | 1/10 |
| 4 | 2 | DIBAL-H (2) | 85 | 1/16 | |
| 5 |  | 2 | DIBAL-H (2) | 89 | 1/3.5 |
| 6 |  | 2 | DIBAL-H (2) | 99 | 1/2 |
| 7 |  | 2 | DIBAL-H (2) | 74 | 1/4 |
| 8 |  | 2 | DIBAL-H (2) | 71 | 1/3 |
| Run | Substrates | Reagent (equiv) | Conditions (°C, h) | Yield (%) | Ratio |
|---|---|---|---|---|---|
| 1 |  | BuLi (1) | −78, 0.25 | 76 | 1/1.9 |
| 2 | MAD (1)/BuLi (1) | −78, 0.3 | 41 | 1/1.4 | |
| 3 | MAPH (1)/BuLi (1) | −78, 0.3 | 76 | 1/6.5 | |
| 4 | MAPH (2)/BuLi (1) | −78, 0.3 | 45 | 1/14 | |
| 5 |  | MAPH (1)/BuLi (1) | −78, 0.5 | 80 | 1/4.3 |
| 6 | MAPH (2)/BuLi (1) | −78, 0.5 | 60 | 1/10.8 | |
| 7 |  | MAPH (1)/BuLi (1) | −78, 0.5 | 60 | 1/2.9 |
| 8 | MAPH (2)/BuLi (1) | −78, 1 | 58 | 1/24 | |
| 9 |  | MAPH (1)/BuLi (1) | −78, 0.5 | 90 | 1/2.7 |
| 10 | MAPH (2)/BuLi (1) | −78, 1 | 68 | 1/56 |
Finally, we would like to discuss the excellent report of Tsuji and colleagues.34) Tsuji and colleagues recently discovered a novel method for carrying out catalytic hydrosilylation reactions that are selective for more sterically hindered ketones. These workers observed that CuCl/tBuONa and the highly bulky bowl-shaped phosphane (bsp) ligand (Chart 21) comprise a catalytic system that can be used to promote selective hydrosilylation reactions of typically poorly reactive bulky ketones with Ph2SiH2 in the presence of their non-bulky counterparts (Table 13). The key feature driving the selectivity of this process is found in the aggregation/disaggregation processes that occur via interactions of the copper complexes with ketones35,36) (Chart 22). Both ketones and aldehyde reversibly form complexes with the copper hydride species ligated to the phosphine. However, owing to the bulkiness of the bsp ligands and bulky alkoxide moieties, complexes formed from bulky ketones have lower nuclearity and, consequently, extremely high reactivities with the silane. In contrast, complexes derived from less hindered ketones or aldehydes generate tighter complexes that have lower reactivities.
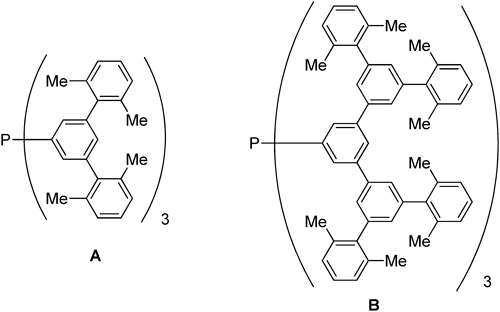
 | |||
|---|---|---|---|
| Run | Substrates | Ligand | Yield of alcohol (%) |
| 1 |  | B | 81/12 |
| 2 |  | B | 90/5 |
| 3 | PPh3 | 8/96 | |
| 4 |  | B | 92/9 |
| 5 |  | B | 72/21 |
| 6 |  | B | 88/2 |
| 7 | A | 87/3 | |
| 8 |  | B | 99/11 |
| 9 |  | B | 89/11 |
| 10 | PPh3 | 21/79 | |
| 11 |  | B | 71/17 |

Research in the area of in situ protection over a more than 30 year period has led to the discovery of a number of methods to carry out reverse-reactivity selective reactions of carbonyl compounds. However, applications in organic synthesis have lagged behind the rapid development of these techniques. Moreover, a large number of carbonyl group reactions, including reductive aminations and α-substitutions, have not been probed in the context of the existing in situ protection methodologies, which for the most part have only been explored in the framework of reduction and/or alkylation processes. In addition, the hydrosilylation protocol described by Tsuji et al. is the only catalytic method developed for this purpose. As a consequence of the fact that in situ protection represents a potentially general strategy to control the chemoselectivity of organic transformations, we believe that intense interest will continue in this area and that significant developments will be made that foster the science of organic synthesis.